In der modernen Chemie und Materialwissenschaft sind präzise simulationsbasierte Methoden essentiell, um neue Moleküle und Materialien zu entwerfen und deren Eigenschaften vorherzusagen. Insbesondere bioorganische Systeme erfordern aufgrund ihrer Komplexität und Größe eine detaillierte Betrachtung atomarer Wechselwirkungen. Traditionelle quantenchemische Verfahren, insbesondere jene, die auf der Lösung der Schrödingergleichung beruhen, bieten zwar eine hohe Genauigkeit, sind jedoch massiv in ihrer Skalierbarkeit eingeschränkt. Die Rechenzeit wächst meist exponentiell mit der Zahl der betrachteten Atome, sodass sie für viele praktische Anwendungen unattraktiv oder gar unbrauchbar werden. Genau hier setzen neuronale Netzwerkpotentiale (NNPs) an und verändern die Landschaft der chemischen Simulationen grundlegend.
Neuronale Netzwerkpotentiale fungieren als schnelle Approximationen quantenmechanischer Berechnungen. Indem sie große Mengen an hochwertigen Referenzdaten aus Dichtefunktionaltheorie (DFT) oder höherwertigen Methoden lernen, sind sie in der Lage, Energiezustände und Kräfte in molekularen Systemen mit nahezu gleicher Genauigkeit vorherzusagen, allerdings zu einem Bruchteil der Rechenzeit. Trotz dieser vielversprechenden Eigenschaften weisen viele existierende Modelle noch Einschränkungen hinsichtlich Genauigkeit, Übertragbarkeit und Geschwindigkeit auf, die ihre praktische Anwendbarkeit einschränken. Mit Egret-1 wurde nun ein neuer Meilenstein in der Entwicklung von NNPs vorgestellt. Dieses Modell basiert auf der MACE-Architektur – einer leistungsfähigen neuronalen Netzstruktur, die speziell dafür entwickelt wurde, atomare Umgebungen detailliert zu erfassen und komplexe chemische Wechselwirkungen zu modellieren.
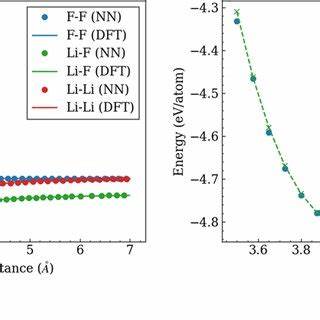
Egret-1 ist ein vortrainiertes Modell, dessen Trainingsdaten diverse chemische Umgebungen aus den Bereichen Hauptgruppenelemente, organische Moleküle und biomolekulare Systeme umfassen. Diese breite Abdeckung macht es zu einem äußerst vielseitigen Werkzeug für Wissenschaftler verschiedenster Disziplinen. Eine der beeindruckendsten Eigenschaften von Egret-1 ist seine Genauigkeit im Vergleich zu klassisch eingesetzten quantenchemischen Methoden. In verschiedenen Testszenarien – darunter Torsionswinkel-Scans, das Ranking von Konformeren sowie die Geometrieoptimierung – erzielt Egret-1 gleiche oder sogar bessere Ergebnisse als etablierte Methoden. Dabei bleibt der Rechenaufwand um mehrere Größenordnungen geringer, was simulationsintensive Anwendungen erheblich beschleunigt.
Die Open-Source-Verfügbarkeit von Egret-1 trägt maßgeblich zur Verbreitung und Weiterentwicklung im wissenschaftlichen Umfeld bei. Forscher können das Modell frei nutzen, anpassen und in ihre eigenen Simulationstools integrieren, was die Innovationsgeschwindigkeit in der Material- und Moleküldesignforschung deutlich erhöht. Gleichzeitig fördert dies die Transparenz und Reproduzierbarkeit wissenschaftlicher Arbeiten, ein zunehmend wichtiges Kriterium in der modernen Forschung. Die Effizienz von Egret-1 resultiert nicht nur aus der zugrunde liegenden Architektur, sondern auch aus der sorgfältigen Auswahl und dem Umfang der Trainingsdaten. Durch das Einbinden vielfältiger chemischer Umgebungen wurden die Modelle so trainiert, dass sie generalisierbar sind und verschiedene molekulare Systeme robust und akkurat abbilden können.
Dieser Aspekt ist besonders in der bioorganischen Chemie von Bedeutung, bei der molekulare Flexibilität und komplexe Wechselwirkungsmuster entscheidend für Funktionen und Wirkungen sind. Trotz dieser bemerkenswerten Fortschritte weist die Arbeit rund um Egret-1 auch auf bestehende Herausforderungen hin, die in zukünftiger Forschung adressiert werden müssen. So sind etwa bestimmte Arten von Elektronenkorrelationen, seltene chemische Reaktionen oder extrem große Molekülsysteme noch immer schwierige Fälle, die aktuelle NNPs nur eingeschränkt abbilden können. Zudem stellt die optimale Erweiterung der Datenbasis und die Feinabstimmung der Netzwerkarchitektur einen fortlaufenden Entwicklungsprozess dar, um die Modelle noch universeller und zuverlässiger zu machen. Die praktische Anwendung von Egret-1 erstreckt sich über zahlreiche Wissenschaftsgebiete.
In der Wirkstoffentwicklung können schnellere und genauere Simulationen dabei helfen, vielversprechende Moleküle gezielt zu identifizieren und zu optimieren, ohne aufwändige experimentelle Testreihen. Auch in der Materialwissenschaft ermöglicht die verbesserte Simulation von atomaren Strukturen und deren Dynamik die gezielte Entwicklung neuartiger funktioneller Materialien, etwa für Energie- oder Nanotechnologieanwendungen. Zusätzlich eröffnet die Möglichkeit, Egret-1 flexibel in bestehende Simulationsplattformen zu integrieren, vielen Forschungsteams den Zugang zu modernster Technologie ohne signifikante Kosten oder Barrieren. Diese Demokratisierung der Simulationstechnologie wird voraussichtlich zu einer erheblichen Beschleunigung der wissenschaftlichen Entdeckungsprozesse führen. Ein weiterer Vorteil von Egret-1 liegt in seiner Skalierbarkeit.
Die Kombination aus hoher Rechengeschwindigkeit und Genauigkeit bedeutet, dass selbst umfangreiche molekulare Systeme und Simulationen mit langem Zeitverlauf handhabbar werden. Dies überschreitet die Grenzen klassischer DFT-Methoden bei weitem und ermöglicht neue Forschungsansätze, die zuvor aufgrund von Ressourcenmangel nicht realisierbar waren. Die Entwicklung von Egret-1 ist ein klares Beispiel dafür, wie künstliche Intelligenz und maschinelles Lernen selbst komplexe Naturphänomene modellierbar machen können. Dabei wird deutlich, dass interdisziplinäre Zusammenarbeit zwischen Chemikern, Informatikern und Physikern der Schlüssel zur Erschließung neuer wissenschaftlicher Horizonte ist. Die Weiterentwicklung solcher Modelle wird künftig eine wichtige Rolle bei der Lösung drängender Herausforderungen in Medizin, Umwelt und Materialtechnik spielen.
Zusammenfassend lässt sich sagen, dass Egret-1 nicht nur ein technologischer Fortschritt im Bereich der Simulation bioorganischer Systeme ist, sondern auch eine fundamentale Veränderung im Ansatz der computergestützten Chemie darstellt. Seine Fähigkeit, DFT-ähnliche Genauigkeit mit enormer Geschwindigkeit zu kombinieren und dabei offen verfügbar zu sein, setzt neue Standards für zukünftige Forschung und Anwendung. Die Forschungsgemeinschaft wartet gespannt darauf, wie Egret-1 in den kommenden Jahren weiterentwickelt wird, welche neuen Funktionen und Verbesserungen implementiert werden und welchen Einfluss es langfristig auf Wissenschaft und Industrie haben wird. Durch die zunehmende Bedeutung von präzisen, schnellen und zugänglichen Simulationswerkzeugen ist Egret-1 zweifellos ein Meilenstein auf dem Weg zu einer nachhaltigen, effizienten und innovativen Chemie der Zukunft.