Die Chemie als Wissenschaftsdisziplin befindet sich im stetigen Wandel, getrieben von Innovationen in Verfahren, Materialien und Technologien. Mit der digitalen Transformation und dem Aufkommen künstlicher Intelligenz (KI) zeichnen sich neue Wege ab, um komplexe Herausforderungen effizienter zu bewältigen. Ein besonders faszinierender Fortschritt sind die großen Sprachmodelle (Large Language Models, LLMs), die durch Feinabstimmung auf spezifische chemische Fragestellungen heute bereits in der Lage sind, präzise Vorhersagen zu treffen und Forschungsprozesse zu beschleunigen. Große Sprachmodelle wurden ursprünglich darauf trainiert, Sprache zu verstehen und zu generieren, indem sie riesige Mengen allgemeiner Textdaten aus dem Internet lernen. Diese Grundausbildung ermöglicht es ihnen, vielfältige Aufgaben rund um Textverständnis und -produktion zu erledigen.
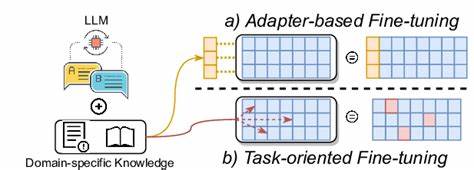
Anders als klassische Machine-Learning-Modelle für Chemie benötigen LLMs keine vorab definierte Merkmalsvektorisierung, sondern können natürliche Sprache oder chemische Repräsentationen wie SMILES direkt verarbeiten. Dies eröffnet vor allem chemisch weniger erfahrenen Forschern neue Möglichkeiten, KI-gestützte Analysen durchzuführen. Die Forschung hat gezeigt, dass das Feinabstimmen dieser vortrainierten Modelle auf chemiespezifische Datensätze hervorragende Resultate erzielt, selbst wenn die verfügbaren Datenmengen vergleichsweise gering sind. Dabei werden aus den ursprünglichen großen Modellen spezialisierte Varianten, die auf konkrete Fragen wie Vorhersage von Materialeigenschaften, Reaktionsverläufen oder Syntheseerfolgen trainiert sind. Die Leistung solcher Modelle wird häufig mit traditionellen Methoden wie Random Forest oder XGBoost verglichen – und die LLMs erweisen sich meist als ebenbürtig oder sogar überlegen.
Ein zentraler Vorteil der Feinabstimmung von LLMs liegt darin, dass chemische Systeme in natürlicher Sprache oder standardisierten Textformaten wie SMILES codiert werden können. Beispielsweise kann ein Polymer als Sequenz von Zeichen („A“ und „B“) beschrieben werden, deren Interpretation vom Modell direkt gelernt wird. Die Umwandlung in numerische Merkmale entfällt, sodass der komplexe Prozess der feingliedrigen Featurisierung, der viel Expertenwissen erfordert, überflüssig wird. Daraus resultiert eine einfachere und zugänglichere Schnittstelle zwischen Chemie und künstlicher Intelligenz. Die Anwendungsfälle sind breit gestreut.
So konnte an polymere Haftenergien anhand ihrer Sequenz vorhergesagt werden, mit einer Genauigkeit, die konventionelle Modelle übertraf. Ähnlich überzeugend sind die Ergebnisse bei der Prognose von Materialeigenschaften wie Schmelzpunkten, dynamischer Viskosität oder der Phasentrennung von Proteinen. Letzteres ist besonders bemerkenswert, da Proteinsequenzen sehr lang und komplex sind, LLMs jedoch auch hier mit minimaler Vorverarbeitung hochpräzise Klassifizierungen ermöglichen. Auch komplexe Anwendungen in der Materialwissenschaft profitieren von der Flexibilität der Modelle. Die Prognose der Mikrostruktur von Magnesiumlegierungen oder das Vorhersagen von Nanopartikelstrukturen aus Röntgenstreudaten zeigen, dass feinabgestimmte LLMs in der Lage sind, mit heterogenen, unvollständigen oder langkettigen Datensätzen umzugehen und dabei hohe Genauigkeiten zu erzielen.
Damit bieten sie Forschern effektive Tools zur Reduktion experimenteller Aufwände und Simulationen. Bei der Reaktionsvorhersage zeigen LLMs ebenfalls große Stärken. So können Aktivierungsenergien für organische Reaktionen oder Wirkungsgrade für katalytische Prozesse zuverlässig eingeschätzt werden. Besonders praktisch ist die Möglichkeit, selbst bei kleinen Datensätzen, wie bei kinetischen Polymerisationsscreenings, mit wenigen Anpassungen akzeptable Vorhersagen zu erhalten. Dies erleichtert nicht nur die Planung von Experimenten, sondern auch die systematische Optimierung von Syntheseprotokollen.
Die Leistungsfähigkeit der Modelle geht jedoch über molekulare Fragestellungen hinaus. Feinabgestimmte LLMs können auch komplexe Systeme und Anwendungen modellieren, wie beispielsweise Gasaustausch in Metall-organischen Gerüstverbindungen (MOFs), Thermodesalinierung oder die Stabilität von Gassensoren. In solchen multidimensionalen Settings bietet die natürliche Sprache als Eingabe die Möglichkeit, sowohl qualitative als auch quantitative Variablen flexibel zu kombinieren und so umfassende prädiktive Modelle zu erzeugen. Neben den zahlreichen Erfolgen zeigen sich auch klare Limitationen und Herausforderungen. Die Leistungsfähigkeit der Modelle hängt stark von der Verfügbarkeit und Ausgewogenheit der Datensätze ab.
In der Chemie ist es häufig so, dass erfolgreiche Experimente überproportional berichtet werden, während Misserfolge selten dokumentiert sind. Diese Verzerrung führt zu unausgewogenen Trainingsdaten, was die Vorhersagequalität insbesondere für seltene, aber relevante „Top-Performer“ beeinträchtigt. Strategien wie schrittweises Verengen des Zielbereichs sowie gezielte Datensammlung können hier Abhilfe schaffen. Die Frage der Repräsentation ist ein weiterer wichtiger Faktor. Während standardisierte Formate wie SMILES sich gut bewähren, kann das Hinzufügen zusätzlicher strukturbezogener Merkmale nicht immer deutliche Verbesserungen bringen.
Das bedeutet, dass die sorgfältige Auswahl und Kombination von Repräsentationen für konkrete Fragestellungen sinnvoll bleibt. Ein markantes Ergebnis der Untersuchungen ist, dass es keine signifikanten Unterschiede in der Vorhersagequalität zwischen unterschiedlichen LLM-Architekturen gab. Modelle wie GPT-J-6B, Llama-3.1-8B und Mistral-7B konnten vergleichbare Ergebnisse liefern, was darauf hindeutet, dass es primär auf die Qualität der Feinabstimmung und die Datenbasis ankommt. Die Nutzung von LLMs in der Chemie bringt auch praktische Vorteile mit sich.
Die einfache Formulierung von Fragen und Eingaben in natürlicher Sprache erleichtert die Zusammenarbeit zwischen Forschungsteams unterschiedlicher Disziplinen. Darüber hinaus sinkt die Hürde für Nicht-Experten, KI-gestützte Analysen einzusetzen, was die Integration solcher Techniken in den wissenschaftlichen Alltag beschleunigt. Zukünftige Forschungsrichtungen könnten die Verknüpfung von LLMs mit hochauflösenden quantenchemischen Berechnungen umfassen, um Synenergien zwischen rechnerischer Genauigkeit und Datenverfügbarkeit zu nutzen. Ebenso verspricht die grössere Offenlegung von negativen Experimenten und fehlgeschlagenen Synthesen eine stärkere Datenbasis für die Ausbildung dieser Modelle. Insgesamt bietet die Feinabstimmung großer Sprachmodelle ein mächtiges Werkzeug, um die Komplexität chemischer Systeme besser zu verstehen und Vorhersagen zu treffen, die Forschung und Entwicklung effizienter gestalten.
Die Möglichkeit, mit geringer Datendichte brauchbare Prognosen zu erhalten, und die einfache Einbindung natürlicher Sprache sind Schlüsselelemente für die breite Akzeptanz und den Einsatz dieser Technologie in der chemischen Praxis. Mit der fortschreitenden Entwicklung von KI-Technologien und einer verbesserten Dateninfrastruktur könnten feinabgestimmte LLMs bald zu einem unverzichtbaren Bestandteil chemischer Forschungs- und Innovationsstrategien werden – von der Moleküldesignphase bis hin zur Prozessoptimierung in der Industrie. Die Digitalisierung der Chemie erhält hiermit einen mächtigen Impuls, der über traditionelle Methoden hinaus neue Horizonte eröffnet.