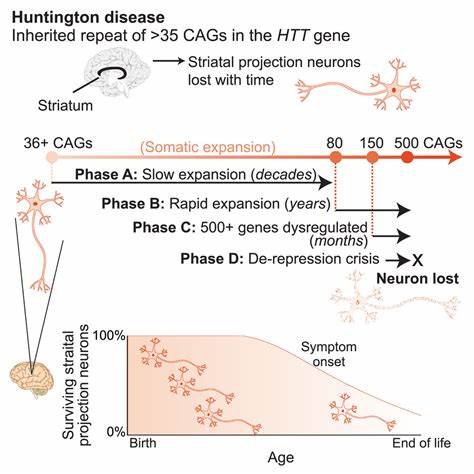
Huntington-Krankheit (HD) ist eine verheerende neurodegenerative Erkrankung, die durch eine pathologische Expansion von CAG-Trinukleotid-Wiederholungen im HTT-Gen verursacht wird. Diese genetische Mutation führt zu einer anormalen Verlängerung von Polyglutamin (Poly-Q) in dem Huntingtin-Protein, was schließlich zum Absterben von Neuronen und zum Fortschreiten motorischer, kognitiver und psychiatrischer Symptome führt. Trotz intensiver Forschung bleibt bisher eine kausale Therapie aus, was die Suche nach neuartigen Ansätzen zur Intervention bei der molekularen Ursache von HD unabdingbar macht. Einer der wesentlichen Faktoren für den Krankheitsverlauf ist die Somatische Instabilität dieser CAG-Wiederholungen. Nach der Geburt kann es im Körper zu einer dynamischen Änderung der Länge dieser Repeats kommen, insbesondere in neuronalem Gewebe, wo sich diese Bereiche im Lauf der Zeit weiter verlängern können.
Diese sogenannte somatische Expansion korreliert stark mit der Frühmanifestation der Krankheit und deren Schweregrad, was die Bekämpfung oder Verhinderung dieser Expansionen zu einem potenziell wirksamen therapeutischen Ziel macht. In avantgardistischer Forschung wurde kürzlich eine innovative Form des Gen-Editings – das sogenannte Basen-Editing – dazu genutzt, um gezielt einzelne Nukleotidbasen innerhalb der CAG-Trinukleotid-Wiederholungen zu verändern. Dabei werden cytosin- oder adeninbasierte Änderungen an den betroffenen Abschnitten vorgenommen, die die homogener Struktur der Wiederholungen unterbrechen und eine Stabilisierung des Genoms bewirken sollen. Im Gegensatz zu klassischen Genscheren-Methoden wie CRISPR/Cas9, die Doppelstrangbrüche verursachen, ermöglichen diese Baseneditoren präzise, punktuelle Korrekturen ohne DNA-Strangbrüche und damit mit einem reduzierten Risiko von unerwünschten Mutationen. Forscher konnten in Patienten-primärzellen zeigen, dass durch gezielte Einfügen sogenannter CAA-Unterbrechungen in ansonsten rein aus CAG bestehenden Wiederholungssequenzen nicht nur die Anzahl der somatischen Expansionen reduziert wird, sondern sich die Repeat-Länge sogar verkürzen ließ.
Diese natürlichen Unterbrechungen kommen vereinzelt auch bei Menschen vor, die trotz längerer CAG-Tracts milder oder später erkranken. Die gezielte Erzeugung dieser Unterbrechungen durch Basen-Editing simuliert somit eine protektive natürliche Variante. Erste Anwendung dieser Methode in Mausmodellen, die humanisierte HTT-Gene mit pathologisch langen CAG-Repeats tragen, hat die hohe Effizienz und Nachhaltigkeit der Baseneditierung in den Zellen des zentralen Nervensystems bestätigt. Nach intraventrikulärer Injektion eines AAV9-basierten Vektors, der den Baseneditor und die spezifische Führungs-RNA trägt, konnten bis zu zwei Drittel der HTT-Allele erfolgreich geändert werden. Die Folge war eine signifikante Reduzierung der CAG-Expansionen in Nervenzellen der Maus-Hirnrinde und des Striatums, den besonders betroffenen Regionen bei HD.
Bemerkenswert ist, dass die Intervention nicht nur das Fortschreiten der Expansion aufhält, sondern auch eine leichte Kontraktion der schädlichen Wiederholungen bewirkt. Aufgrund der Verbindung zwischen Länge der CAG-Trains und Krankheitsausprägung eröffnet dies einen vielversprechenden neuen Weg, um sowohl Krankheitsentstehung als auch Progression zu beeinflussen. Neben der hohen Effizienz überzeugte die Baseneditierung durch ein günstiges Sicherheitsprofil. Ausgedehnte ganze-genomische Untersuchungen zeigten zwar potenzielle Off-Target-Aktivitäten, jedoch traten diese überwiegend in nicht-codierenden Bereichen des Genoms auf oder resultierten in stillen Mutationen, die keine Veränderung im Proteincodierungsschema bedingen. Trotz einiger weniger nonsynonymer Änderungen waren die meisten vorhersagbar biologisch verträglich, wobei unerwünschte Auswirkungen bisher nicht beobachtet wurden.
Diese Studien erlauben einen bedeutenden Einblick in das bislang wenig verstandene Zusammenwirken von Wiederholungsunterbrechungen und genomischer Stabilität. Sie bestätigen, dass nicht nur die Länge des gesamten CAG-Tracts entscheidend ist, sondern auch die Anwesenheit von Unterbrechungen, welche die DNA-Struktur verändern und somit DNA-Reparaturprozesse vor expansionsfördernden Fehlern schützen. In der klinischen Perspektive sind die Möglichkeiten von Baseneditierungstechnologien vielfältig. Vergleichbar wurden ähnliche Ansätze bei der Friedreich-Ataxie untersucht, bei der pathogene GAA-Trinukleotid-Expansionen im FXN-Gen die Krankheit verursachen. Die Installation von Unterbrechungen auch in dieser Gen-Region hat zu einer Verbesserung der FXN-Expression und Reduktion der Repeat-Instabilität geführt, was auf eine allgemeine Anwendbarkeit des Konzepts bei verschiedenen trinukleotidbedingten Erkrankungen hinweist.
Die Auslieferung durch AAV9-Vektoren ist sowohl in der Hirnforschung als auch generell in der Gentherapie etabliert. Diese Vektoren bieten aufgrund ihrer Neuronotropie und Langzeitexpression einen idealen Träger für Genom-editierende Werkzeuge, insbesondere wenn frühzeitige Interventionen im Entwicklungsstadium möglich sind. Dennoch müssen mögliche Immunantworten und die dauerhafte Expression von Editoren kritisch beobachtet werden, um langfristige Sicherheit gewährleisten zu können. Ein zukünftiger Entwicklungsschritt wird die Verfeinerung der Baseneditoren sein, um deren Spezifität weiter zu erhöhen und unerwünschte Nebeneffekte weiter zu reduzieren. Zudem könnten alternative Lieferwege, wie virale Vektorvarianten mit breiterem Zelltypspektrum oder nicht-virale Methoden wie Virus-ähnliche Partikel, die zeitlich begrenzte Expression ermöglichen, weitere Vorteile bieten.
Aus therapeutischer Sicht bergen diese Fortschritte die Chance, die bislang unheilbaren Folgen der Huntington-Krankheit in ihrer molekularen Ursache zu begegnen. Werden somatische Expansionen effektiv unterbunden, lassen sich Krankheitsbeginn verzögern, Symptome mindern oder sogar Prävention betreiben, bevor irreversible neuronale Schäden entstehen. Die Erkenntnisse öffnen zudem Wege zur genetischen Modulation bei weiteren polyglutaminbedingten Krankheiten und anderen TNR-basierten Neurodegenerationen, von Ataxien bis Spinal- und Bulbär-Muskulärer Atrophie. Durch die Möglichkeit, spezifische Wiederholungsstrukturen zu verändern, wird eine personalisierte Medizin greifbarer, bei der genetische Variationen gezielt beeinflusst werden können. Abschließend lässt sich festhalten, dass die Anwendung von Baseneditierung zur Reduktion von Huntington-assoziierten CAG-Wiederholungsexpansionen einen Meilenstein in der Erforschung von TNR-Erkrankungen darstellt.
Durch präzise genetische Eingriffe in patienteneigenen Zellen und relevanten Tiermodellen erbringen diese Ansätze überzeugende Belege für ihr therapeutisches Potenzial. Die Weiterentwicklung und klinische Erprobung könnten zukünftig das Leben von Patienten mit Huntington und verwandten Krankheiten maßgeblich verbessern und neue Standards in der Behandlung genetischer Neurologischer Erkrankungen setzen.