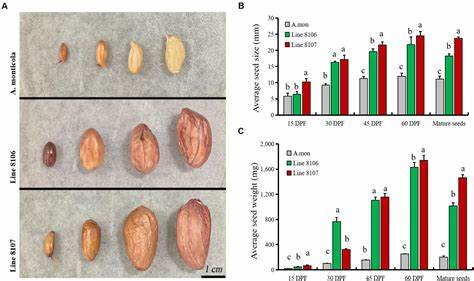
Die Erdnuss (Arachis hypogaea L.) zählt zu den bedeutendsten ölhaltigen und nahrhaften Leguminosen weltweit. Insbesondere die Saatgutgröße und das Gewicht sind Schlüsselfaktoren, die den Ertrag und die Qualität dieser Kulturpflanze maßgeblich beeinflussen. Trotz der wirtschaftlichen und ernährungsphysiologischen Bedeutung der Erdnuss sind die molekularen Grundlagen, die diese wichtigen agronomischen Merkmale steuern, bislang nur unzureichend erforscht. Neuerdings ermöglicht die Pangenomanalyse, also das Zusammenführen genetischer Informationen mehrerer Varianten einer Art, einen ganzheitlichen Blick auf die genomische Vielfalt und ihre funktionellen Auswirkungen.
Hierbei werden insbesondere auch größere strukturelle Variationen erforscht, die sich als bedeutende Quelle für phänotypische Unterschiede herausstellen. Der Ursprung der kultivierten Erdnuss geht auf eine natürliche Hybridisierung zweier wildlebender diploider Arten, Arachis duranensis (A-Untergenom) und Arachis ipaensis (B-Untergenom), vor mehreren tausend Jahren zurück. Daraus entstand ein allotetraploides Genom (AABB), das im Verlauf der Domestikation sich durch selektive Züchtung von kleinen Wildsamen zu größeren, ertragreicheren Sorten entwickelte. Diese Domestikationsprozesse führten jedoch zu komplexen genomischen Veränderungen, die bislang schwer zu entschlüsseln waren, nicht zuletzt wegen der Polyploidität und der hohen Repetitivität des Erdnussgenoms. Moderne genomische Methoden, kombiniert mit einer breit gefächerten Stichprobe von 269 Erdnuss-Accessions, darunter Wildtypen, Landrassen und verbesserten Sorten, erlauben nun erstmals einen tiefgreifenden Vergleich der genetischen Unterschiede.
Die dabei entstehende Pangenomstruktur identifiziert über 50.000 Genfamilien, die sich in Kern-, Verteilte und Private Familien gliedern. Bemerkenswert ist die ungleiche Verteilung von sogenannten strukturellen Variationen (SVs) im A- und B-Untergenom, wobei das A-Untergenom eine höhere Frequenz aufweist. Solche Variationen umfassen größere genetische Veränderungen wie Insertions- oder Deletionsereignisse, die das Genom reorganisieren und Funktionen beeinflussen können. Eine bedeutende Entdeckung ist ein 275-Basenpaar großes Deletionsereignis im Gen AhARF2-2.
Dieses Gen kodiert einen Auxin-Response-Faktor, der insbesondere das Wachstum und die Entwicklung von Samen reguliert. Die Deletion führt zum Verlust einer Interaktionsdomäne mit weiteren regulatorischen Proteinen (AhIAA13 und TOPLESS), was die Hemmung von AhGRF5 verringert – einem Wachstum fördernden Faktor. Das Resultat daraus ist eine gesteigerte Samenexpansion und somit größere Saatgutkörper. Dieses Beispiel illustriert eindrucksvoll, wie strukturelle Genvarianz direkt zu physiologischen Veränderungen und damit zu agronomisch relevanten Merkmalen führen kann. Darüber hinaus wurden über 1.
300 SVs identifiziert, die mit der Domestikation in Verbindung gebracht werden können, sowie nahezu 200 Variationen, die spezifisch mit Saatgutgröße und Gewicht korrelieren. Solche Erkenntnisse sind für die Züchtung von erheblichem Wert, da sie präzise molekulare Marker und Zielgene für gezielte Manipulationen bereitstellen. Die Bedeutung dieser Genvariationen spiegelt sich auch in der Expressionsanalyse wider, die bestätigt, dass SVs innerhalb von Promotor- und Exonregionen die Genaktivität modulieren und somit die phänotypische Ausprägung beeinflussen können. Neben AhARF2-2 liefert die Studie weitere interessante Einblicke in Gene wie AhCKX6, dessen Variation in der 3'-UTR mit einer Verringerung der Genexpression und einer Erhöhung der aktiven Cytokininspiegel einhergeht. Cytokinine sind Pflanzenhormone, die Zellteilung und Wachstumsprozesse fördern.
Damit ist AhCKX6 ein Kandidatgen, das durch genetische Variationen eine Schlüsselrolle bei der Bestimmung der Samenentwicklung spielt. Die Pangenomanalyse fördert nicht nur das Verständnis der genetischen Basis für die Saatgutgröße, sondern zeigt auch die komplexe Evolution und asymmetrische Domestikation zwischen den beiden Untergenomen der Erdnuss. Während das A-Untergenom stärker von SVs geprägt ist, weist das B-Untergenom im Verlauf der Züchtung spezifische Selektionssignale auf. Dieses ungleiche Selektionsmuster spiegelt sich auch in funktionellen Genfamilien wider, etwa in der Anreicherung von Krankheitsresistenzgenen im B-Untergenom, die eine Balance zwischen Ertrag und Abwehrmechanismen unterstützen. Die Verfügbarkeit hochwertiger Referenzgenomen, kombiniert mit graph-basierten Methoden zur SV-Detektion und Genotypisierung, ermöglicht eine detaillierte Abbildung der genetischen Landschaft.
Damit entsteht eine zuverlässige Datengrundlage für weitere funktionelle Studien und Züchtungsprogramme. Insbesondere die Kombination von Assembly-basierten und Read-basierten SV-Erkennungen verbessert die Auflösung und Genauigkeit deutlich gegenüber früheren Ansätzen. Ein wichtiger praktischer Aspekt ist, dass viele der identifizierten SVs in Verbindung mit bekannten oder potenziellen Domestikationsgenen stehen, welche die größten Beiträge zur Variation bei der Saatgutgröße leisten. Dieser genetische Schatz kann genutzt werden, um mithilfe moderner Markerassistierter Selektion oder Genome Editing Verfahren Züchtungsfortschritte nachhaltiger und schneller zu erzielen. Gleichzeitig erlaubt das Verständnis der genomischen Architektur der Erdnuss eine bessere Erhaltung der genetischen Diversität und stärkt die Widerstandsfähigkeit gegenüber biotischen und abiotischen Stressfaktoren.
Zusammengefasst bietet die Pangenomanalyse der Erdnuss einen Meilenstein zur Aufschlüsselung der genetischen Mechanismen, die hinter der Quantitativen Variation der Samenmerkmale stehen. Die gewonnenen Daten liefern nicht nur wertvolles Wissen zur pflanzengenetischen Forschung im Allgemeinen, sondern stellen eine unverzichtbare Ressource dar für die gezielte Optimierung einer global bedeutsamen Kulturpflanze. Sie ebnet den Weg für eine nachhaltige Steigerung von Ertrag und Qualität der Erdnuss in zukünftigen Züchtungsprogrammen auf Basis molekularbiologischer und genomischer Werkzeuge.