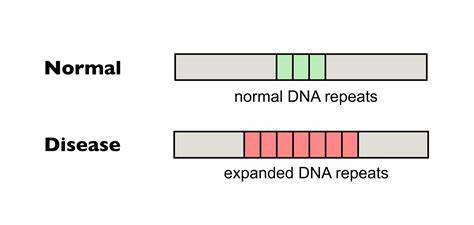
Die Huntington-Krankheit stellt eine der komplexesten neurodegenerativen Erkrankungen dar und ist durch eine abnormale Expansion von CAG-Wiederholungen im Huntingtin-Gen (HTT) charakterisiert. Diese repetitiven Sequenzen kodieren für Polyglutamin-Peptide, deren Verlängerung im Protein die Krankheitsentstehung und -progression maßgeblich beeinflusst. Insbesondere ist die somatische Expansion dieser Wiederholungen in neuronalen Zellen eng mit dem Ausbruch und der Verschlechterung der Symptome verbunden. Ein innovativer Lernzweig in der biomedizinischen Forschung beschäftigt sich derzeit mit der gezielten Bearbeitung dieser DNA-Wiederholungen, um ihre weitere Expansion zu verhindern oder sogar zurückzudrängen. Dabei nimmt die Basen-Editierung von trinukleotidalen Wiederholungen einen zentralen Stellenwert ein und verspricht neue therapeutische Möglichkeiten, die bislang unerreichbar schienen.
Die Huntington-Krankheit wird autosomal-dominant vererbt und tritt meist nach dem 30. bis 50. Lebensjahr auf. Die individuelle Dauer bis zum Krankheitsausbruch und der Schweregrad sind stark von der Anfangslänge der CAG-Trinukleotid-Wiederholungen abhängig. Längere Wiederholungssequenzen korrelieren typischerweise mit einem früheren Beginn und rasanterem Krankheitsverlauf.
Bemerkenswert ist, dass sich diese CAG-Sequenzen somatisch, also innerhalb der Zellen des betroffenen Patienten, weiter verlängern können. Das bedeutet, dass mit fortschreitendem Alter der Betroffenen die Wiederholungen insbesondere in Zellen des zentralen Nervensystems wachsen, was zur Verstärkung neurodegenerativer Prozesse beiträgt. Die somatische Instabilität von Wiederholungen ist daher ein entscheidender Faktor, der sowohl die Pathogenese als auch den Therapieverlauf beeinflusst. Traditionelle therapeutische Maßnahmen zielen bisher meist auf die Symptomlinderung oder die Verzögerung der Krankheitsprogression ab, während die genetische Ursache der CAG-Expansion praktisch kaum angegangen werden konnte. Neurowissenschaftliche Revolutionen wie die CRISPR-Cas-Technologie und insbesondere die Entwicklung von Basen-Editoren eröffnen hier neue Behandlungsperspektiven.
Im Gegensatz zu den klassischen Genom-Editing-Methoden, die Doppelstrangbrüche im DNA-Strang verursachen und durch unvorhersehbare Reparaturmechanismen Folgeeffekte riskieren, ermöglichen Basen-Editoren präzise und kontrollierte Nukleotidveränderungen auf einzelner Basenebene. Diese Apparate können gezielt einzelne Cytosin-Basen in Thymin und Adenin in Guanin umwandeln – ein Mechanismus, der insbesondere genutzt werden kann, um innerhalb der CAG-Trinukleotidsequenz gezielte Interruptionsmutationen einzuführen. Natürlicherweise wurden in Patienten mit stabileren oder mild verlaufenden Formen der Krankheit sogenannte CAA-Unterbrechungen innerhalb der repetitiven CAG-Sequenzen identifiziert. Diese Unterbrechungen bestehen aus ähnlichen kodierten Aminosäuren, führen jedoch zu einer erhöhten Stabilität der DNA-Repeats und verhindern signifikant deren somatische Expansion. Studien deuten darauf hin, dass selbst einzelne CAA-Interruptionsstellen die Krankheitsausprägung um mehr als ein Jahrzehnt hinauszögern können.
Dieses Verständnis hat zur Theorie geführt, dass eine gezielte Einführung solcher Unterbrechungen mittels Basen-Editierung therapeutisch nutzbar sein könnte. Aktuelle bahnbrechende Studien zeigen, dass Cytosin-Baseneditoren (CBEs) verwendet werden können, um genau solche CAA-Interruptionsmutationen in CAG-Trinukleotidrepeats der HTT-Gene von Patienten- und Modellzellen einzufügen. In Experimenten mit patienteneigenen Fibroblasten wurde nachgewiesen, dass durch die Einbringung von Interruptionsbasen die somatische Expansion deutlich reduziert und in einigen Fällen sogar eine Wiederholungsreduktion beobachtet werden kann. Langzeitstudien in Zellkulturen belegen, dass diese editierte Sequenz stabil bleibt und eine weitere schädliche Erweiterung verhindert wird. Besonders beeindruckend sind auch In-vivo-Experimente in Mouse-Modellen der Huntington-Krankheit, in denen die CAG-Unterbrechungen durch AAV9-vermittelte Lieferung der Basen-Editoren in das zentrale Nervensystem injiziert wurden.
Es konnte gezeigt werden, dass durch frühzeitige Behandlung eine signifikante Verringerung der somatischen CAG-Expansionen im Gehirn erreicht wird, insbesondere in Schlüsselregionen wie Cortex und Striatum, die für die motorischen Defizite der Krankheit verantwortlich sind. Die Effizienz der Transduktion erreichte in einigen Fällen über 70 % editierter Huntingtin-Allele in neuronalen Zellen, was als großer Schritt in der therapeutischen Genom-Editierung gilt. Darüber hinaus zeigen vergleichbare Ansätze bei der Friedreich-Ataxie, einer weiteren durch trinukleotidale Wiederholungsausdehnung verursachten neurodegenerativen Erkrankung, dass Adenin-Basen-Editoren (ABEs) GAA-Repeats im FXN-Gen entsprechend unterbrechen können. Die Strategie, die Wiederholung zu „brechen“, ähnelt dabei der Vorgehensweise bei Huntington, was die Vielseitigkeit und das große Potenzial der Methode unterstreicht. Ein zentrales Anliegen der Forschung ist die Evaluierung von möglichen Off-Target-Effekten, also unbeabsichtigten Editierungen an anderen genomischen Stellen, die zu potenziellen Nebenwirkungen oder gar Schäden führen könnten.
Um diesem Aspekt gerecht zu werden, wurden breit angelegte Genomanalysen mittels CIRCLE-seq und Hochdurchsatz-Sequenzierung durchgeführt, die zeigen, dass die meisten Off-Target-Effekte bei repeat-ähnlichen Sequenzen auftreten, wobei viele dieser Editierungen entweder keine Proteine kodieren oder zu synonymer Mutation führen, also den Aminosäurecode nicht verändern. Die Ergebnisse weisen darauf hin, dass das Risiko signifikanter unerwünschter Veränderungen für eine klinische Anwendung beherrschbar sein könnte, insbesondere wenn optimierte Editor-Versionen und gezielte Verabreichungsformen verwendet werden. Sicher ist, dass die Genom-Editierung mittels Basen-Editoren den attraktiven Vorteil besitzt, die pathogenen Wiederholungen auf molekularer Ebene direkt und dauerhaft zu modifizieren – ein Aspekt, den symptomorientierte Therapien nicht erreichen können. Deshalb eröffnet dieses Verfahren nicht nur Möglichkeiten für therapeutische Interventionen, sondern auch für tiefere Einblicke in die molekulare Biologie von Trinukleotidwiederholungskrankheiten. Herausforderungen bleiben dennoch bestehen.
Die Verbesserung der Effizienz und Spezifität der Basen-Editoren, die Optimierung der viralen Vektorlösungen für eine zielgerichtete und sichere Genomeinbringung sowie die Erweiterung der Erkenntnisse in adulten und humanrelevanten Geweben sind aktuelle Forschungsprioritäten. Derzeit erfolgen die meisten Anwendungen noch im Neonatalstadium in Tiermodellen, während die Translation in die klinische Therapie bei erwachsenen Patienten noch auf sich warten lässt. Des Weiteren ist das biologische Verhalten der repeat-expansion in verschiedenen Zelltypen innerhalb des zentralen Nervensystems noch nicht vollständig verstanden. Beispielsweise spielen auch Gliazellen und andere nicht-neuronale Zelltypen, ebenso wie periphere Gewebe, eine wichtige Rolle im Krankheitsgeschehen. Deshalb werden künftige Studien sich bemühen müssen, die Gen-Editierung auf möglichst viele betroffene Zelltypen auszuweiten, um die umfassende therapeutische Wirkung zu gewährleisten.
Insgesamt stellt die gezielte Basen-Editierung von trinukleotidalen Wiederholungen ein vielversprechendes neues Kapitel der Huntington-Krankheit und verwandter Erkrankungen dar. Indem sie somatische Expansionen hemmt und die schädlichen DNA-Tracts stabilisiert oder gar verkürzt, könnte diese innovative Strategie letztlich das Fortschreiten der Krankheit verlangsamen, symptomatische Erleichterungen schaffen oder sogar die Lebensqualität der Betroffenen verbessern. Das Engagement in dieser Forschungsrichtung erfordert interdisziplinäres Wissen aus Molekularbiologie, Genetik, Neurowissenschaften und klinischer Medizin. Die rasante technologische Weiterentwicklung der Genome-Editing-Werkzeuge sowie deren Kombination mit gerätegestützter Präzisionsmedizin versprechen eine spannende Zukunft, in der bislang unheilbare genetische Krankheiten aktiv und sicher behandelt werden können. Zukunftsweisende Klinische Studien werden zeigen müssen, inwieweit solche molekularen Eingriffe langfristig sicher und effektiv sind.
Bis dahin bilden präklinische Erfolge und die stetige Verfeinerung der Basen-Editierung eine solide Basis für einen potenziellen Durchbruch in der Behandlung der Huntington-Krankheit und ähnlicher neurodegenerativer Erkrankungen.