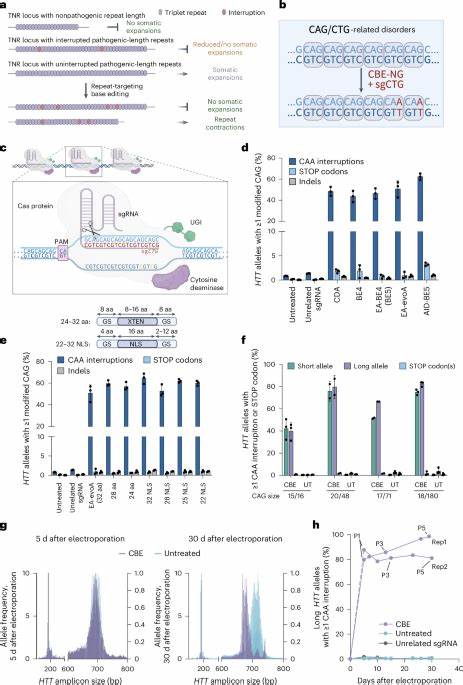
Die Huntington-Krankheit (HD) zählt zu den sogenannten trinukleotid Repeat-Erkrankungen, einer Gruppe von neurologischen Störungen, die durch die pathologische Erweiterung repetitiver DNA-Sequenzen in bestimmten Genen verursacht werden. Diese Krankheiten zeichnen sich durch eine fortschreitende Degeneration von Nervenzellen aus und weisen bisher keine Heilung auf. Ein besonders kritischer Aspekt bei HD ist die sogenannte somatische Repeat-Expansion der CAG-Wiederholungen im Huntingtin-Gen (HTT), die über die Lebensdauer von Patienten zu einer exponentiellen Verschlechterung der Erkrankung führen kann. Moderne medizinische Forschung konzentriert sich daher zunehmend auf Strategien, diese Instabilität im Genom gezielt zu beeinflussen und auszubremsen, um den Krankheitsverlauf zu verlangsamen oder gar zu stoppen. Die somatische Instabilität beschreibt das Phänomen, dass sich die Anzahl der CAG-Wiederholungen in den Nervenzellen eines Patienten im Laufe der Zeit erhöht.
Diese Erweiterung korreliert direkt mit dem Ausbruch und der Schwere der Huntington-Erkrankung. Je länger die Wiederholungsstrecke, desto stärker die Toxizität des Huntingtin-Proteins und desto schneller schreitet die neurodegenerative Symptomatik voran. Dies hat die Forschung vor die Herausforderung gestellt, Mechanismen zum Schutz vor somatischen Erweiterungen zu finden. Innovative Ansätze im Bereich der Genom-Editierung, insbesondere die sogenannten Base-Editing-Technologien, ermöglichen es heute, gezielt einzelne Nukleotide innerhalb der CAG-Trinukleotid-Sequenzen zu verändern, ohne dabei das gesamte Genom destabilisieren zu müssen. Im Gegensatz zu klassischen Methoden der Genom-Editierung, die auf Doppelschnittstellen der DNA abzielen, erfolgt die Base-Editierung präzise und ohne Doppelstrangbrüche.
Dies minimiert potenzielle unerwünschte Nebenwirkungen wie Chromosomenbrüche oder unerwünschte Mutationen. Im Zentrum dieser innovativen Technologie stehen Cytosin- und Adenin-Base-Editoren, die gezielt spezifische Basensubstitutionen durchführen. So lassen sich im Fall von Huntington-affinen CAG-Wiederholungen einzelne CAG-Trinukleotide gezielt in CAA-Trinukleotide umwandeln. Diese CAA-Sequenzen gelten als natürliche Unterbrechungen in der ansonsten monotonen CAG-Kette und sind in der Bevölkerung bereits als stabilisierende Varianten bekannt. Studien zeigen, dass solche Interruptions im DNA-Strang die Entstehung von höheren DNA-Strukturkomplexen verhindern, welche für unerwünschte Expansionen verantwortlich sind.
Die Einführung solcher Unterbrechungen durch Base Editing verringert also nachweislich die somatische Instabilität in Genen, die von Trinukleotid-Expansionen betroffen sind. In verschiedenen Zellkulturmodellen sowie in Patientenzellen mit Huntington-Syndrom wurde bereits demonstriert, dass durch gezielte Einfügung von CAA-Interruptions im HTT-Gen signifikant weniger somatische Erweiterungen beobachtet werden. Die Technologie nutzt eigens designte Single-Guide-RNAs (sgRNAs), die spezifisch an die CAG-wiederholenden Sequenzen binden und dort die Base-Editoren anleiten. Durch präzise C-to-T-Substitutionen kann so eine gesunde Variation der Wiederholungsregion erzeugt werden. Nicht nur in Patientenfibroblasten, sondern auch in präklinischen Tiermodellen wurden diese Interventionen erfolgreich getestet.
Im Htt.Q111-Mausmodell, das eine humane Huntingtin-Mutation mit einer ausgeprägten CAG-Expansion enthält, führte die Auslieferung von Base-Editoren mittels adeno-assoziierter Viren (AAV9) in das Zentralnervensystem zu einer effizienten Umwandlung der CAG-Wiederholungen in CAA und somit zu einer deutlichen Reduktion der somatischen Repeat-Expansion. Dies konnte über einen Beobachtungszeitraum von mehreren Wochen bis Monaten verifiziert werden und zeigte, dass die Basis-Editierung nicht nur die Dynamik der Expansion entschleunigt, sondern auch wiederholt zu einer Verkürzung der nicht-pathogenen Trinukleotid-Anzahl führte. Diese Entwicklungen eröffnen einen therapeutisch äußerst wichtigen Ansatz, da die somatische Instabilität als ein zentraler Treiber für den Spätbeginn und die rapide Verschlechterung bei Huntington gilt. Die Verlängerung der Lebensphase, in der die Expansionen gering bleiben, könnte daher das aufkeimende neurodegenerative Fortschreiten dramatisch verzögern.
Zudem wurde beobachtet, dass die Umwandlung der CAG in CAA-Trinukleotide eine stille Mutation erzeugt, die den Proteincode nicht ändert, was die Sicherheit der Intervention erhöht und Nebenwirkungen potenziell minimiert. Zusätzlich zur HTT-Locus wurde die Base-Editing-Technologie auch auf andere durch Trinukleotid-Expansionen verursachte Erkrankungen mit Erfolg übertragen. Beispielsweise bei Friedreich-Ataxie, einer weiteren schweren neuromuskulären Erkrankung, die durch die Expansion von GAA-Trinukleotiden im FXN-Gen bedingt ist, können durch spezifische Adenin-Base-Editoren GAA-Wiederholungen sicher und effizient in stabile Varianten umgewandelt werden. Im Vergleich zur bislang undifferenzierten therapeutischen Behandlung stellt dies einen entscheidenden Fortschritt in der personalisierten Gen-Therapie dar. Die größte Herausforderung bei der Base-Editing-Technologie besteht derzeit jedoch in der Minimierung möglicher Off-Target-Effekte.
Wissenschaftler arbeiten daran, die Präzision der Editors zu erhöhen, sodass nur die gewünschten Sequenzen verändert werden, ohne dass unerwünschte Mutationen in anderen Genen entstehen. Derzeitige Studien belegen aber, dass viele der Off-Target-Editierungen entweder nicht in protein-kodierenden Bereichen auftreten oder nur synonyme, also nicht proteinverändernde Mutationen hervorrufen. Zudem sind mögliche Auswirkungen solcher Nebenwirkungen im Zusammenhang mit der Therapie von neurodegenerativen Erkrankungen bislang gering einzuschätzen, speziell im Hinblick auf die lebensverlängernden Vorteile. Neben der genetischen Kontrolle der somatischen Repeat-Expansionen erforschen Wissenschaftler auch die molekularen Mechanismen, die für die Instabilität verantwortlich sind. Dabei spielt die fehlerhafte DNA-Reparatur, insbesondere der Einfluss des Mismatch-Repair-Systems, eine zentrale Rolle.
Die Basenunterbrechungen und dadurch veränderte Struktur des repetitiven Genabschnitts verhindern offenbar die Bildung von DNA-Schleifen, die sonst DNA-Reparaturprozesse fehlerhaft auslösen. Dies ist ein positiver Nebeneffekt der Base-Editing-Strategie. Das therapeutische Potential dieser Ansätze ist enorm, auch wenn die Verabreichung von Genombearbeitungswerkzeugen in den menschlichen Körper weiterhin mit Hürden verbunden ist. Die Verwendung von AAV9-Vektoren ermöglicht eine effiziente Expression insbesondere in neuronalen Zellen, die bei Huntington stark betroffen sind. Die gezielte, möglichst frühe Intervention – idealerweise noch vor Auftreten symptomatischer Beschwerden – könnte somit den Krankheitsverlauf dramatisch verlängern oder möglicherweise verhindern.
Darüber hinaus stellen die Erkenntnisse um die präventive Wirkung von Interruptions in Trinukleotid-Wiederholungen eine neue Dimension im Verständnis der Pathophysiologie dar. Stabile Unterbrechungen in den genetischen Repeat-Sequenzen scheinen bislang unterschätzte pharmakogenetische Mechanismen zu sein, die Einfluss auf Alter, Progression und Auftreten neurodegenerativer Erkrankungen haben. Die Kombination aus High-Precision-Genom-Editierung und immer weiter verbesserten viralen Vektor-Systemen dürfte in den nächsten Jahren eine Revolution in der Behandlung von Huntington und verwandten Krankheiten auslösen. Klinische Studien müssen diese vielversprechenden Vorab-Ergebnisse validieren und die langfristige Sicherheit der Gen-Editierung beurteilen. Dennoch zeichnet sich heute klar ab, dass die gezielte Reduktion somatischer Repeat-Expansionen auf molekularer Ebene ein erfolgversprechender Weg ist, um die tödliche Huntington-Krankheit dauerhaft zu beeinflussen.
Abschließend zeigt die Forschung eindrucksvoll, wie durch die Nutzung natürlicher genetischer Varianten und modernster Biotechnologien das Erbgut genetisch belasteter Patienten so modifiziert werden kann, dass schädliche Mutationsdynamiken gebremst werden. Diese Fortschritte unterstreichen die Rolle der personalisierten Medizin und der Genom-Editierung als Schlüsseltechnologien für zukünftige neurodegenerative Therapien mit dem Ziel, das Leben von Betroffenen nachhaltig zu verbessern.








