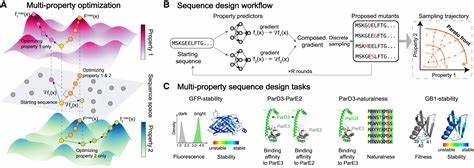
Die Entwicklung neuer Proteine mit spezifischen Eigenschaften stellt eine der spannendsten Herausforderungen im Bereich der Biotechnologie und Bioinformatik dar. Die Möglichkeiten, Proteine gezielt so zu designen, dass sie mehrere Anforderungen gleichzeitig erfüllen, eröffnen neue Wege für die Medikamentenentwicklung, Biokatalyse sowie für die Erzeugung innovativer Biomaterialien. Dabei stellt die gleichzeitige Berücksichtigung multipler Zielgrößen – wie Bindungsaffinität, Stabilität, Löslichkeit und Spezifität – höchste Ansprüche an die Designmethoden und erfordert besonders flexible und effiziente Optimierungsansätze. Ein vielversprechender Fortschritt in diesem Bereich bietet das sogenannte funktionale Mehrziel-Protein-Design mittels kontinuierlicher Relaxation, das die kombinierte Anwendung verschiedener maschinell gelernter Modelle sowie differentieller Optimierungsverfahren ermöglicht. Dieses Verfahren stellt eine innovative Lösung für die hohe Komplexität des proteinsequenz- und strukturbezogenen Suchraums dar und nutzt kontinuierliche Optimierungsmethoden, um diskrete Proteinsequenzen effizient zu gestalten.
Das fundamentale Problem beim Protein-Design ist die extrem große Anzahl möglicher Aminosäuresequenzen. Ein einzelnes Protein mit nur 50 Aminosäuren besitzt bereits 20 hoch 50 Möglichkeiten, was einer enormen Suchraumgröße entspricht. Die Diskretheit des Problems erschwert klassisches Optimieren erheblich und macht naive Ansätze wenig praktikabel. Gleichzeitig ist die Bewertung jeder Sequenz durch simulationsgestützte Methoden oder experimentelle Validierung zeitintensiv und teuer. Kontinuierliche Relaxation löst dieses Problem, indem die diskrete Sequenzwahl auf einen kontinuierlichen Wahrscheinlichkeitsraum überführt wird.
Statt eine einzelne Aminosäure festzulegen, wird jede Position als Wahrscheinlichkeitsverteilung über alle 20 Aminosäuren dargestellt. Dadurch kann auf diesem kontinuierlichen Raum mit Gradientenverfahren optimiert werden, was deutlich effizienter ist als diskrete MCMC-Methoden oder evolutionäre Algorithmen. Dieser Ansatz nutzt die Tatsache, dass viele neuronale Netzwerke zur Proteineigenschaftsvorhersage eine „one-hot“-Sequenzkodierung zu einem kontinuierlichen Repräsentationsvektor verarbeiten. Indem man die Eingabe von festen Einsen auf Wahrscheinlichkeiten verteilt, wird ein differenzierbares Loss-Funktional geschaffen, das mithilfe moderner Gradient-basierter Optimierer kontinuierlich minimiert werden kann. Ein zentraler Vorteil dieser Methodik ist die einfache Erweiterbarkeit und Modularität der Zielfunktionen.
Mehrere, teils tief lernende Modelle – etwa für die Bindungsaffinität an ein Zielprotein, Vorhersage von Off-Target-Bindung, Proteinexpression in Bakterien und Löslichkeit – können flexibel kombiniert werden. Dadurch entsteht ein zusammengesetztes Verlustmaß, das die gewünschten Eigenschaften simultan berücksichtigt und so zu funktionell optimierten Proteinsequenzen führt. Die eingesetzten Modelle reichen von strukturprädiktiven Algorithmen wie Boltz-1 oder Boltz-2, Alphafold2, über Inverse-Folding-Modelle wie ProteinMPNN bis hin zu Protein-Sprachmodellen wie ESM (Evolutionary Scale Modeling) und weiteren Regressionsmodellen, die beispielsweise Stabilität vorhersagen. Jedes dieser Modelle liefert differenzierbare Verlustterme, die sich leicht zu einer gemeinsamen Zielfunktion verschmelzen lassen. Die Optimierungsverfahren basieren überwiegend auf proxilem Spiegelabstieg mit entropischer Regularisierung, was einer generalisierten proximalen Gradientenmethode entspricht.
Diese Algorithmen fördern die Konvergenz zu spitzen Verteilungen auf dem Wahrscheinlichkeits-simplex, die am Ende auf proteintypische, diskrete Sequenzen abgebildet werden können. Weitere Möglichkeiten sind projizierter Gradientenabstieg oder Methoden, die ein Softmax-Ramping verwenden, um die Sequenzdiskretisierung sukzessive zu verstärken. Mit dieser Methodik lassen sich spezifische Aufgaben angehen, wie beispielsweise die Entwicklung von Peptid-Bindern, die eine hohe Affinität an ein Zielprotein aufweisen, aber gleichzeitig kaum unerwünschte Bindung an andere ähnliche Proteine eingehen. Ein gleichzeitiges Optimieren der Expressionsfähigkeit in Bakterienzellen sorgt dafür, dass die entworfenen Sequenzen praktisch herstellbar sind. Die Berücksichtigung der Löslichkeit verhindert Aggregation und Stabilitätsprobleme.
Die Implementierung solcher Systeme erfolgt häufig in modernen Frameworks wie JAX, die Just-in-Time-Kompilierung, automatische Differenzierung und GPU-Beschleunigung integrieren. Das erlaubt schnelle Experimente mit verschiedensten Verlustkombinationen und ermöglicht den Einsatz maßgeschneiderter Optimierer, die für die jeweilige Problemstellung ideal angepasst sind. Ein weiterer starker Vorteil liegt in der Erlaubnis, benutzerdefinierte Verlustterme einzufügen. Diese können auf bestehenden Frameworks aufsetzen, um beispielsweise cysteinfreie Designs zu erschaffen, die keine unerwünschten Disulfidbrücken bilden, oder um Fitnessfunktionen aus Simulationen oder evolutionären Ansätzen als differentiable Module zu integrieren. Diese Flexibilität eröffnet neue Möglichkeiten für die zielgerichtete Entwicklung von Protein-Domänen mit anspruchsvollen funktionellen Eigenschaften.
Trotz des enormen Potenzials stehen diese Methoden jedoch noch vor Herausforderungen. Die Handhabung der hyperparametrischen Abstimmung, die Sicherstellung der biophysikalischen Realisierbarkeit der Designs und die Vermeidung von adversarialen Sequenzen – die nur in silico gut abschneiden, aber in vitro kaum brauchbar sind – erfordern sorgfältige Validierung und zusätzliche Filterverfahren. Es gibt derzeit weder umfangreiche experimentelle Verifizierungen noch standardisierte Benchmarks, die eine eindeutige Bewertung erlauben. Nichtsdestotrotz spiegeln diese Entwicklungen den aktuellen Stand der Protein-Designforschung wider, der traditionelle Methoden mit maschinellem Lernen und differenzierbarer Programmierung verbindet. Die modulare Natur der Softwarearchitekturen erleichtert die Integration zukünftiger Fortschritte, wie zum Beispiel die Einbindung ligandenspezifischer Verlustterme, erweiterter Optimierungsalgorithmen wie differenzierbarer Monte-Carlo-Verfahren oder Ressourcen-effizienter serieller Verlustberechnung zur Vermeidung von Speicherengpässen.
Zukunftsgerichtet wird mit einer noch stärkeren Einbindung von Protein-Sprachmodellen und generativen Modellen gerechnet, die direkt aus sequenziellen Eingaben neue Designs ableiten können, gesteuert durch differentielles Feedback von Eigenschaftsprädiktoren. Ebenso erscheint die Kombination aus kontinuierlicher Relaxation und diskreten Optimierungsstrategien vielversprechend, da so die Stabilität und Qualität der finalen Sequenzlösungen verbessert werden kann. Im Bereich der Biotechnologie sowie der Pharmaindustrie kann dieses Protein-Design-Verfahren maßgeblich dazu beitragen, die Entwicklung neuer Therapeutika oder funktionaler Biomoleküle zu beschleunigen. Die Fähigkeit, Designs punktgenau auf verschiedene Eigenschaften abzustimmen, ohne aufwändige iterative Laborexperimente, spart Zeit und Kosten. Insgesamt repräsentiert die kontinuierliche Relaxation im Mehrziel-Protein-Design einen paradigmatischen Wandel hin zur hochskalierbaren, modularen und datengetriebenen Gestaltung biomolekularer Systeme.
Die Herausforderung bleibt, Algorithmen und Modelle so zu verfeinern und in die praxisrelevante Pipeline zu integrieren, dass sie biologische Komplexität und experimentelle Realität optimal abbilden. Die enge Zusammenarbeit von Computerwissenschaftlern, Biologen und Chemikern wird daher entscheidend sein, um das volle Potenzial dieser neuartigen Technologie auszuschöpfen und innovative Proteine mit maßgeschneiderter Funktionalität zu realisieren.








