Die Erforschung der menschlichen Herkunft ist eine der faszinierendsten Herausforderungen der Wissenschaft. Während archäologische Funde und genetische Untersuchungen bereits eine Vielzahl an Erkenntnissen über die Evolution des Homo sapiens liefern, bringt die neuste Forschung mit einem strukturierten Koaleszenzmodell nun auch innovative Einsichten über die tiefen Wurzeln unserer Vorfahren. Dieses Modell offenbart eine komplizierte, gemeinsame Abstammungsstruktur, die weit zurück in die Menschheitsgeschichte reicht und alle heutigen Menschen verbindet. Unter den verschiedenen Methoden der Populationsgenetik hat die Koaleszenz-Theorie eine zentrale Rolle eingenommen. Sie beschreibt, wie sich genetische Linien durch Generationen zurückverfolgen lassen, um gemeinsame Vorfahren zu identifizieren.
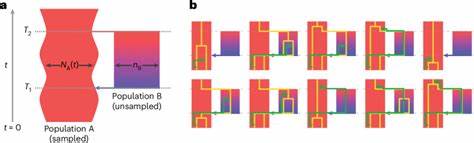
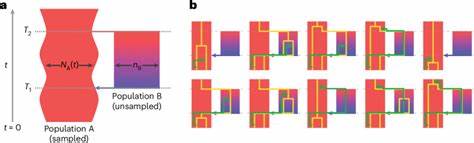
Traditionelle Koaleszenzmodelle gehen oftmals von einer panmiktischen Population, sprich einer zufällig paarenden Gruppe, aus. Doch reale Populationen weisen häufig eine komplexere Struktur auf, beispielsweise durch Unterteilungen, wiederholte Trennungen und Vermischungen, die bei der üblichen Annahme fehlinterpretiert werden können. Hier setzt das strukturierte Koaleszenzmodell an: Es integriert explizit die Tatsache, dass sich Populationen vor langer Zeit aufspalten und später wieder miteinander verschmelzen können. Die jüngste Studie, veröffentlicht im renommierten Fachmagazin Nature Genetics, nutzt dieses Modell, um die Geschichte moderner Menschen auf einer völlig neuen Ebene zu rekonstruieren. Die Analyse berücksichtigt dabei zwei ancestral getrennte Populationen, die sich vor etwa 1,5 Millionen Jahren abzweigten und sich ungefähr 300.
000 Jahre später wieder vermischten. Diese Vermischung wird mit einem Intensitätsverhältnis von etwa 80 zu 20 Prozent zwischen den beiden Gruppen modelliert. Die Bedeutung dieser Entdeckung ist vielfältig. Zum einen beweist sie, dass das Erbgut heutiger Menschen tief in einer doppelt verzweigten und wieder vereinten Population wurzelt. Das widerspricht der vereinfachten Vorstellung einer einzigen, zusammenhängenden menschlichen Vorfahrenpopulation und unterstützt Theorien, die eine komplexere, strukturierte Evolution in Afrika und darüber hinaus nahelegen.
Die Population mit dem größeren Anteil, sogenannte Population A, erlebte unmittelbar nach der Aufspaltung eine dramatische Flaschenhalsepisode, das heißt, ihre genetische Vielfalt schrumpfte stark. Gleichzeitig war Population B über einen langen Zeitraum größer und stabiler. Die Forscher konnten anhand genetischer Daten und dem neuen Modell zudem Abschnitte im Genom von heute lebenden Menschen identifizieren, die von jeder der beiden ursprünglichen Populationen stammen. Dabei zeigt sich, dass genetisches Material aus der Minderheitslinie B in bestimmten Regionen des Genoms tendenziell seltener vorkommt, speziell in der Nähe von codierenden Genabschnitten. Dies weist darauf hin, dass diese Allele möglicherweise nachteilig und daher durch Selektion negativ beeinflusst wurden, wenn sie auf dem Hintergrund der Hauptpopulation auftauchten.
Eine weitere bemerkenswerte Erkenntnis des Modells ist der Bezug zu den Neandertalern und Denisovanern. Die genetischen Bereiche, die der Mehrheitspopulation A zugeordnet sind, weisen eine stärkere Nähe zu diesen archaischen Menschen auf, was darauf hindeutet, dass die Population A auch der primäre genetische Vorfahre für diese Gruppen gewesen sein könnte. Demgegenüber scheint die Bevölkerung B genealogisch weiter entfernt zu sein. Das strukturierte Koaleszenzmodell namens cobraa, das hinter dieser Arbeit steckt, erweitert bestehende Verfahren erheblich. Es kombiniert die Hidden-Markov-Modellierung mit einer fein abgestimmten Darstellung von Populationstrennung und -vermischung und berücksichtigt dabei die Rekombination im Genom.
Die Anwendung auf umfangreiche Datensätze wie dem 1000-Genomes-Projekt und dem Human Genome Diversity Project ermöglicht eine präzise Einordnung zeitlicher Ereignisse in der menschlichen Abstammungslinie. Die Forschung verdeutlicht auch, dass traditionelle Modelle wie das PSMC, welche vollständige Panmixia annehmen, wichtige Aspekte wie eine frühe Populationsteilung und fluktuierende Populationsgrößen nicht oder nur unzureichend abbilden können. Die neuen Erkenntnisse über die tiefgreifende Struktur machen deutlich, dass wir die menschliche Evolution als ein viel komplexeres Geflecht verstehen müssen, in dem verschiedene Populationen über lange Zeit getrennt existierten und sich später wieder vermischten. Darüber hinaus liefert das Modell interessante Einsichten in die Evolution anderer Tierarten, wenn sein Einsatz beispielsweise auf Fledermäuse, Delfine oder Menschenaffen ausgeweitet wird. Dabei liefert es unterschiedlich stark ausgeprägte Hinweise auf vergangene Populationsteilungen und -vermischungen mit teils sehr jungen oder sehr alten Zeitpunkten, was den Charakter der jeweiligen Spezies reflektiert.
Die Auswirkungen dieser Ergebnisse sind weitreichend. Sie verändern unsere Perspektive auf die Ursprünge aller modernen Menschen grundlegend und liefern eine neue Erklärung für die genetische Diversität, die wir heute beobachten. Die Erkenntnisse legen nahe, dass die Evolution unserer Art von kontinuierlicher Trennung, Isolation und späterer erneuter Vermischung geprägt war. Dies spiegelt möglicherweise komplexe Wanderungen, Umweltveränderungen und soziale Dynamiken wider, die im Laufe von Hunderttausenden von Jahren das Bild der menschlichen Evolution formten. Ein weiterer spannender Aspekt ist, dass das Modell hilft, genetische Regionen zu identifizieren, welche bei der Anpassung an veränderte Umweltbedingungen oder zur Vermeidung schädlicher genetischer Kombinationen eine Rolle spielten.
Die signifikanten Korrelationen zwischen genomischen Regionen des Minoritätsanteils und funktionalen Aspekten wie der Nähe zu codierenden Sequenzen oder Bereichen starker Hintergrundselektion verweisen auf selektive Prozesse, die Einfluss auf die heutige genetische Landschaft haben. Zukunftsweisend könnte die Integration von strukturierten Koaleszenzmodellen mit anderen genetischen Analysemethoden, wie der Untersuchung des Allelfrequenzspektrums oder der Rekonstruktion von Stammbäumen anhand ganzer Genome, noch tiefere Einblicke in die Evolutionsgeschichte des Menschen ermöglichen. Erscheinen könnten so neue Erkenntnisse über bislang unbekannte archaische Populationen oder über komplexe Migrations- und Vernetzungsmuster. Weiterhin lohnt sich ein Vergleich der modellierten genetischen Ereignisse mit archäologischen Funden. Zum Beispiel stimmen die geschätzten Zeitpunkte der Populationsteilung und -vermischung bemerkenswert gut mit den frühesten bekannten Fossilien von anatomisch modernen Menschen überein, etwa den Funden aus Jebel Irhoud in Marokko.