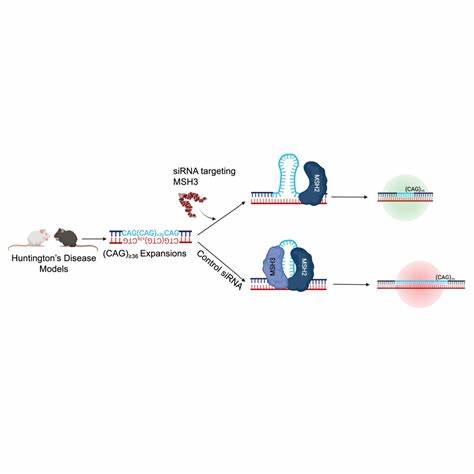
Die Huntington-Krankheit (HD) gehört zu den sogenannten trinukleotid-Repeat-Erkrankungen, bei denen eine abnormale Ausdehnung bestimmter DNA-Wiederholungen im Genom eine zentrale Rolle spielt. Konkret handelt es sich dabei um CAG-Wiederholungen im Huntingtin-Gen (HTT), die bei Betroffenen oft tausendfach verlängert sind und mit fortschreitenden neurologischen Symptomen einhergehen. Doch die Erkrankung ist nicht statisch. Im Laufe des Lebens kann es zu somatischen Repeat-Expansionen kommen, bei denen sich diese CAG-Trinukleotid-Sequenzen in einzelnen Körperzellen weiter vermehren und so den Krankheitsverlauf weiter verschlechtern. Die Möglichkeit, somatische Expansionsprozesse gezielt zu verhindern oder zu reduzieren, könnte daher einen entscheidenden therapeutischen Durchbruch bedeuten.
Die jüngsten Erkenntnisse rund um die Baseneditierung bieten genau hierfür eine vielversprechende Lösung. Baseneditoren sind spezielle Genome-Editing-Werkzeuge, die präzise einzelne Nukleotide umwandeln können, ohne dabei das gesamte DNA-Doppelstrang zu durchtrennen, wie es bei klassischen CRISPR/Cas9-Technologien der Fall ist. Dieses schonendere Verfahren ermöglicht es, selektiv Veränderungen vorzunehmen, die im Fall der Huntington-Krankheit zu einer Stabilisierung der CAG-Wiederholungen führen können. Bei derzeitiger Forschung gibt es eine besondere Fokussierung auf das Einführen sogenannter Unterbrechungen in diese repetitiven Sequenzen. Natürlich vorkommende Basenvarianten wie CAA im CAG-Repeat-Trakt wurden bereits als schützende Faktoren identifiziert, die somatische Instabilität reduzieren und somit den Krankheitsbeginn verzögern können.
Das gezielte Einfügen solcher nicht-pathogener Unterbrechungen durch Baseneditierung kommt demnach einer Genstabilisierung gleich und kann sich als einer der nachhaltigsten Therapieansätze erweisen. Experimentelle Studien an Patientenzellen und Tiermodellen zeigen bereits beeindruckende Erfolge. Dort wurde mit Hilfe von Cytosinbaseneditoren (CBE) eine Umwandlung von CAG zu CAA erreicht, was zu weniger somatischer Erweiterung im HTT-Genabschnitt führt. Die Ergebnisse unterstreichen nicht nur die technische Machbarkeit, sondern auch die klinische Relevanz der Methode. Eine bedeutsame Beobachtung ist, dass die Behandlung dazu beiträgt, die Anzahl der CAG-Repeats über die Zeit nicht nur zu stabilisieren, sondern in bestimmten Fällen sogar zu reduzieren.
Dies deutet auf eine potenzielle Rückbildung schädlicher Genabschnitte hin – ein Meilenstein in der Behandlung genetischer Wiederholungserkrankungen. Ferner wurden antibiotisch zugelassene Vektoren basierend auf Adeno-assoziierten Viren der Serotyp 9 (AAV9) verwendet, um die Baseneditoren gezielt in neuronale Kerngebiete wie den Striatum und die Großhirnrinde zu transportieren, wo die Huntington-Krankheit besonders Schäden verursacht. Die langzeitige Expression der Editoren in den CNS-Tissue führte zu einer nachhaltigen Installation von Unterbrechungen und einer bedeutenden Reduktion der somatischen CAG-Expansion. Neben Huntington identifizieren Wissenschaftler eine parallele Anwendung für Friedreich-Ataxie, eine weitere trinukleotid-Repeat-Erkrankung. Hier sind GAA-Wiederholungen im FXN-Gen beteiligt.
Analog erfolgte der Einsatz von Adeninbaseneditoren (ABE) zur Umwandlung von A•T-Basenpaaren in G•C, um natürliche Unterbrechungen in die GAA-Repeat-Trakte einzubauen. Resultate zeigten eine verbesserte Stabilität und erhöhte Expression von FXN-mRNA, was eine mögliche therapeutische Wirkung andeutet. Zu den wichtigsten Herausforderungen gehört die Minimierung unerwünschter Nebenwirkungen. Zwar sind baseneditierende Verfahren präzise, doch besteht stets die Möglichkeit von Off-Target-Veränderungen, bei denen nicht zielgerichtete Genstellen modifiziert werden. Umfangreiche Analysen der Off-Target-Effekte durch Methoden wie CIRCLE-Seq und Hochdurchsatz-Sequenzierung lassen jedoch darauf schließen, dass viele unerwünschte Veränderungen entweder in nicht-codierenden Bereichen des Genoms stattfinden oder sogar synonyme Mutationen betreffen, die keine Aminosäureänderungen sowie keine Funktionseinschränkungen nach sich ziehen.
Für die klinische Anwendung sind dennoch weitere Sicherheitsbewertungen entscheidend. Insbesondere gilt es, die Auswirkungen auch geringfügiger Off-Target-Editing-Ereignisse in Langzeitstudien sorgfältig zu prüfen und Methoden zur gezielten Reduktion dieser Risiken weiterzuentwickeln. Nicht nur die Technik selbst, sondern auch die Wahl der Vektor-Plattformen spielt eine Rolle. AAV9 ist derzeit die wählbare Option, um zentrale Hirnregionen bei Neugeborenen anzusprechen, doch fehlen therapeutische Zugänge für andere wichtige Gewebe wie Herz oder gliale Zellen, die ebenfalls von der HD pathologisch betroffen sein können. Zukünftige Forschungen richten sich daher darauf, alternative Vektoren oder kombinierte Therapieverfahren zu entwickeln, die auch später im Krankheitsverlauf noch wirksam sind.
Auch wird die Möglichkeit transienter und gezielter Baseneditor-Expression untersucht, etwa über gentechnisch modifizierte Virus-ähnliche Partikel oder mRNA-delivery, um die potenzielle Toxizität durch Dauerexpression zu minimieren. Weitere spannende Forschungsansätze befassen sich zudem mit der Kombination der Baseneditierung mit anderen Therapieformen, etwa RNA-Interferenz oder proteinbasierten Aggregat-Inhibitoren, um synergistische Effekte zu erzielen. Zusammenfassend lässt sich festhalten, dass das gezielte Editieren von Huntington-Repeat-Trakten eine revolutionäre Therapieoption darstellt, die die bisher ungelöste Herausforderung somatischer Repeat-Expansionen adressiert. Die Veränderungen in patienteneigenen Zellen und Tiermodellen belegen das Potenzial, Disease-Modifying-Therapien für eine Gruppe bisher unheilbarer neurodegenerativer Erkrankungen zu etablieren. Die wissenschaftliche Gemeinschaft befindet sich aktuell an einem Wendepunkt, an dem Fortschritte in der Molekulargenetik, Bioinformatik und viralen Vector-Technologie zusammengeführt werden, um die Grundlage für zukünftige klinische Anwendungen zu legen.
Letztlich bieten die vorgestellten Methoden nicht nur Hoffnung für Huntington-Patienten, sondern eröffnen auch einen blueprintartigen Ansatz, der sich auf andere Trinukleotid-Repeat-Erkrankungen übertragen lässt und somit die neurogenetische Therapie von morgen prägen könnte.