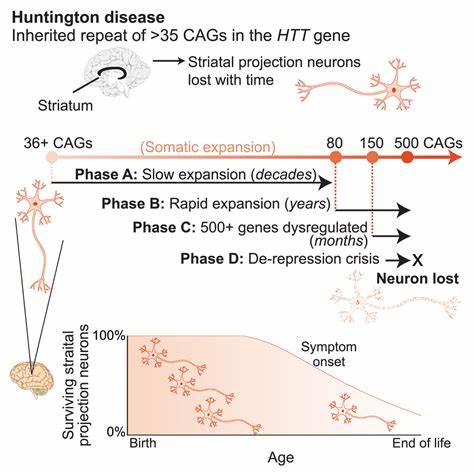
Die Huntington-Krankheit (HD) zählt zu den erblichen neurodegenerativen Erkrankungen, die durch eine abnormale Expansion von trinukleotidalen CAG-Repeat-Sequenzen im HTT-Gen verursacht wird. Diese pathologischen Repeat-Expansionen führen zur Bildung einer toxischen Polyglutamin-Kette im Huntingtin-Protein, welche den neuronalen Zerfall, insbesondere in wichtigen Hirnregionen, begünstigt und sich in fortschreitenden motorischen, kognitiven sowie psychiatrischen Symptomen manifestiert. Während die Krankheit bisher unheilbar ist, zeigen aktuelle wissenschaftliche Fortschritte ein enormes Potenzial in der Bearbeitung dieser genetischen Sequenzen, um den Krankheitsverlauf zu modifizieren und möglicherweise aufzuhalten. Die Instabilität der trinukleotidalen Repeats spielt eine zentrale Rolle bei der Progression der Huntington-Krankheit. Diese repetitive DNA-Sequenz ist nicht nur bei der Entstehung der Mutation relevant, sondern auch bei ihrer somatischen Expansion – einem Prozess, bei dem sich die Anzahl der CAG-Wiederholungen in einzelnen Körperzellen mit der Zeit vergrößert.
Somatische Repeat-Expansionen korrelieren hierbei mit einer schnelleren Krankheitsprogression und einem früheren Symptombeginn. Gerade im zentralen Nervensystem sind diese Veränderungen besonders ausgeprägt und tragen entscheidend zur Neurodegeneration bei. Forscher haben sich deshalb intensiv mit Möglichkeiten beschäftigt, wie man die genomische Instabilität der CAG-Repeats eindämmen kann. Ein vielversprechender Ansatz basiert auf sogenannten Baseneditoren, einer präzisen Form der Genom-editierung, die einzelne Nukleotidveränderungen ohne doppelte DNA-Brüche ermöglicht. Dabei kommen Cytosin- und Adenin-Baseneditoren zum Einsatz, die gezielt bestimmte Basen austauschen können, um störende, rein repetitive Sequenzen zu unterbrechen und dadurch deren Stabilität zu fördern.
Eine wichtige Erkenntnis aus genetischen Studien ist, dass natürliche Unterbrechungen innerhalb der CAG-Repeats, beispielsweise durch gleiche Aminosäure codierende CAA-Kodons, die Stabilität der Repeat-Region erhöhen und die Expansion hemmen. Menschen mit solchen Unterbrechungen zeigen häufig einen verzögerten Krankheitsbeginn oder mildere Symptome. Aufbauend auf diesem Wissen haben Wissenschaftler Cytosin-Baseneditoren (CBE) entwickelt, die gezielt CAG-Tripletts zu CAA umwandeln, um eine solche Schutzwirkung nachzuahmen. In Zellkulturexperimenten wurde gezeigt, dass der Einsatz von CBEs signifikant die Anzahl der störenden Unterbrechungen in den HTT-CAG-Repeats erhöht. Die veränderten Repeat-Sequenzen sind dadurch weniger anfällig für somatische Expansionen.
Besonders nachhaltig ist diese Wirkung in Patientenfibroblasten mit langen pathogenen Repeat-Längen, wo eine Verminderung der Expansion über mehrere Zellteilungen beobachtet werden konnte. Dies unterstreicht das therapeutische Potenzial der Baseneditierung, Repeat-Erkrankungen auf DNA-Ebene entgegenzuwirken. Parallel zu den In-vitro-Studien wurden die baseneditierenden Werkzeuge in Tiermodellen getestet, insbesondere in humanisierten Huntington-Mäusen (Htt.Q111). Durch die Vermittlung einer adeno-assoziierten Virus-Vektortechnologie (AAV9) konnte die Baseneditoren-Botschaft gezielt in relevante Hirnregionen wie Cortex und Striatum eingeschleust werden.
Die Daten zeigten eine effiziente Einführung von CAA-Unterbrechungen in das pathologische CAG-Repeat, begleitet von einer deutlichen und anhaltenden Reduktion der somatischen Repeat-Expansion im Gehirn der behandelten Tiere. Die beobachtete Verhinderung der Repeat-Verlängerung spricht eindeutig für eine protektive Rolle dieser genetischen Modifikation gegen die Krankheitsprogression. Neben Huntington betrifft eine Reihe weiterer neurologischer Erkrankungen eine ähnliche genomische Herausforderung durch in Selbstkopier-Vorgängen verlängerbare Repeat-Sequenzen. Ein Beispiel hierfür ist die Friedreich-Ataxie (FRDA), bei der eine Expansion von GAA-Triplets im FXN-Gen eine progressive Ataxie verursacht. Auch hier wurden Adenin-Baseneditoren (ABE) erfolgreich eingesetzt, um die Repeat-Sequenzen gezielt mit Unterbrechungen zu versehen, die die genomische Instabilität reduzieren.
Analog zu den Ergebnissen bei HD führten die Bearbeitungen in FRDA-Stammzellen sowie in entsprechenden Mausmodellen zu einer durchgehenden Reduktion der somatischen Expansion und einer partielle Wiederherstellung der FXN-mRNA-Expression, die für die Krankheitssymptomatik entscheidend ist. Trotz dieser Erfolge bleibt die Sicherheit und Präzision der Baseneditierung ein wichtiges Forschungsanliegen. Die umfangreiche Analyse von möglichen Off-Target-Effekten zeigte, dass die meisten ungewollten Editierungen in nicht-codierenden Bereichen des Genoms stattfinden und häufig als harmlose, synonym kodierende Mutationen auftreten. Dennoch ist eine sorgfältige Überwachung unerwünschter Veränderungen essenziell, vor allem hinsichtlich potenzieller Auswirkungen auf neuronale Funktionen und langfristige Therapiesicherheit. Die Effektivität und Nachhaltigkeit der Baseneditoren wurde durch das langlebige Expressionsprofil von AAV-Vektoren begünstigt, die dauerhaft vorliegen und somit kontinuierlich DNA-Substrate bearbeiten können.
Dies ermöglicht eine kumulative Anreicherung schützender Unterbrechungen, was langfristig eine stabile Genomstabilität begünstigt. Allerdings stellt eine permanente Expression auch ein Risiko für potenzielle Nebenwirkungen dar, daher sind alternative, zeitlich limitierte Liefermethoden und Editor-Varianten mit reduzierter Off-Target-Aktivität Gegenstand intensiver Forschung. Eine Besonderheit der Baseneditierung bei TNR-Erkrankungen ist, dass die Abfolge der Repeat-Sequenzen selbst vielfältige Zielstellen bietet und damit an zahlreichen Loci potenziell editierbar ist. Die gezielte Unterbrechung der Repeats führt zur Einschränkung der Bildung toxischer, höherer DNA-Strukturen wie Haarnadeln oder Triple-Stränge, die bei ununterbrochenen Repeats somatische Expansionen fördern. Somit kann die Behandlung von somatischen Expansionen ein Türöffner für eine neue Klasse von genetischen Interventionen werden, die nicht nur Huntington, sondern ein breites Spektrum von Repeat-Erkrankungen adressiert.
Der Weg in eine klinische Anwendung bleibt allerdings noch mit Herausforderungen gepflastert. So ist die Distribution des Therapiekonzepts auf alle relevanten Gewebe und Zellen des Menschen, insbesondere in späteren Krankheitsphasen, noch zu optimieren. Die Entwicklung alternativer Vektor-Tropismen, gezielter und kontrollierter Editor-Expression sowie der Ausbau sicherer und effizienten Abgabesysteme werden zentrale Aufgaben für zukünftige Studien sein. Zudem müssen Langzeitstudien sowohl an Tieren als auch in klinischen Studien auf mögliche unvorhergesehene Nebenwirkungen achten. Dennoch markieren die bisherigen Ergebnisse einen Meilenstein in der Huntington- und Friedreich-Ataxie-Forschung.
Sie liefern schlagkräftige Belege dafür, dass genompräzise Baseneditierung gezielt pathogene Repeat-Expansionen in menschlichen Zellen und Modellorganismen korrigieren kann. Damit öffnet sich ein vielversprechendes Fenster zur Prävention oder Verlangsamung neurodegenerativer Krankheiten, bei denen die genetische Grundlage bislang als unveränderlich galt. Werden therapeutische Baseneditierungen zukünftig erfolgreich klinisch einsetzbar sein, könnten Betroffene von Huntington, Friedreich-Ataxie und weiteren trinukleotidalen Repeat-Störungen eine nachhaltige Stabilisierung ihrer genetischen Mutation erleben – ein Paradigmenwechsel in der Behandlung bisher unheilbarer neurodegenerativer Erkrankungen. Die kontinuierliche Erforschung der molekularen Mechanismen somatischer Repeat-Expansionen, kombiniert mit hochpräzisen genetischen Tools, wird die Grundlage für innovative Therapieansätze bilden. Parallel dazu wächst das Verständnis für die genetischen Modifikatoren, die den Krankheitsverlauf beeinflussen.
Die Kombination all dieser Erkenntnisse verspricht, das Schicksal von Patienten mit Huntington-Krankheit und verwandten Erkrankungen grundlegend zu verändern.