Die Huntington-Krankheit (HD) stellt eine der komplexesten genetisch bedingten neurodegenerativen Erkrankungen dar. Sie wird durch eine abnormale Expansion von CAG-Trinukleotid-Wiederholungen im Huntingtin-Gen (HTT) ausgelöst. Diese pathologische Expansion führt zu einer toxischen Veränderung des Huntingtin-Proteins, was letztendlich die Neurodegeneration vor allem in bestimmten Hirnregionen wie dem Striatum und dem Cortex verursacht. Das Fortschreiten der Krankheit führt zu motorischen Einschränkungen, kognitiven Einbußen und psychiatrischen Symptomen, die das Leben der Betroffenen erheblich beeinträchtigen. Trotz intensiver Forschung steht bislang keine kurative Behandlung zur Verfügung, sodass innovative Therapieansätze von großer Bedeutung sind.
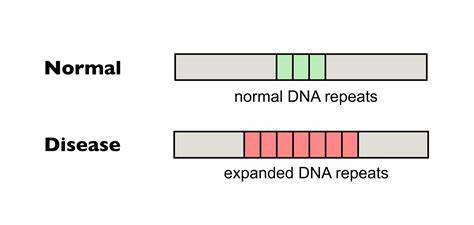
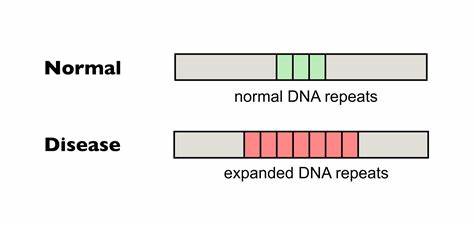
Eines der vielversprechendsten Felder der Huntington-Forschung ist die gezielte Modifikation der trinukleotidischen Repeat-Sequenzen im HTT-Gen mittels modernster Genome-Editing-Technologien. Besonders die somatischen Repeat-Expansionen, also die weiteren Vergrößerungen der CAG-Wiederholungen innerhalb der Körperzellen im Verlauf des Lebens, tragen wesentlich zum Ausbruch und zur Progression der Huntington-Krankheit bei. Somit stellt die Verringerung dieser somatischen Expansionen ein wichtiges Ziel dar. Die kürzlich veröffentlichte Forschungsarbeit zeigt eindrucksvoll, dass präzises Gen-Editing mittels sogenannter Base-Editoren diese schädlichen Wiederholungsausweitungen in Patienten-Zellen und Tiermodellen reduzieren kann. Base-Editing ist eine innovative biotechnologische Methode, die punktgenaue Nukleotidänderungen im Genom ermöglicht, ohne dabei DNA-Doppelstrangbrüche zu induzieren, was die Sicherheit und Effizienz im Vergleich zu älteren CRISPR-Techniken deutlich erhöht.
Im Fokus steht dabei die Umwandlung von reinen CAG-Wiederholungen in Sequenzen, die natürliche Interruptionsmuster aufweisen. Diese sogenannten Interruptions, beispielsweise durch Einfügen von CAA-Codons, sind auf molekularer Ebene synonym und verändern nicht die kodierten Aminosäuren des Huntingtin-Proteins, wirken aber stabilisierend auf die Wiederholungskette. Das Ergebnis ist eine Verringerung der Instabilität der Repeat-Trakte und somit eine signifikante Reduktion von somatischen Expansionen. Mehrere in-vitro-Experimente mit HD-Patientenfibroblasten zeigten, dass die Cytosin-Base-Editoren gezielt einzelne Wiederholungen umwandeln können, was zu einer hohen Rate an Interruptions in den pathogenen CAG-Trakten führte. Bemerkenswert ist, dass die pathogen erweiterten Allele häufiger modifiziert wurden als die normalen, da längere Wiederholungssequenzen mehr Zielstellen für den Editor bieten.
Neben der erfolgreichen Einführung der Interruptions wurde durch Langzeituntersuchungen bestätigt, dass die behandelten Zellen eine verminderte Expansion der Wiederholungen aufweisen, während unbehandelte Kontrollzellen kontinuierlich weitere CAG-Expansionen zeigten. Darüber hinaus erfolgt die Validierung in vivo. In Mausmodellen, die humane HTT-Gene mit pathologisch langen CAG-Trakten tragen, erfolgte die Verabreichung der Base-Editoren durch AAV9-gestützte Vektoren ins zentrale Nervensystem. Nach Behandlung zeigten die Tiere nicht nur eine hohe Effizienz der Interruptions-Einführung in Hirnzellen, sondern auch eine signifikante Verringerung der somatischen Ausdehnungslast der CAG-Wiederholungen in Cortex und Striatum. Neben der Huntington-Krankheit hat die Technologie auch Anwendungspotenzial bei ähnlichen Formen trinukleotidischer Wiederholungserkrankungen, wie beispielsweise der Friedreich-Ataxie, bei der GAA-Repeat-Expansionen im FXN-Gen zu neurodegenerativen Defiziten führen.
Hier konnten Adenin-Base-Editoren eingesetzt werden, um Interruptions innerhalb der GAA-Tracts einzuführen, was den pathogenen Prozess der Wiederholungsausdehnung ebenfalls reduziert. Besonders hervorzuheben ist, dass die Base-Editing-Strategien auf natürlichen sequenzvarianten basieren, die in der Humanpopulation bereits existieren und mit milderen oder verzögerten Krankheitserscheinungen assoziiert sind. Dies bedeutet nicht nur eine effektive Stabilisierung der Wiederholungstrakte, sondern minimiert auch potenzielle Immun- oder Toxizitätsrisiken, die durch de-novo-Mutationen entstehen könnten. Die Off-Target-Analyse der eingesetzten Base-Editoren ergab überwiegend Editierungen in nicht-kodierenden oder synonymen Sequenzbereichen, was für die Sicherheit der Methode spricht. Die Nebenwirkungen auf die genomische Integrität bleiben gering, und die meisten unerwünschten Veränderungen sind entweder unbedenklich oder treten in Genregionen mit geringer Expression im Gehirn auf.
Dennoch ist die fortlaufende Erforschung der Langzeitwirkungen und der möglichen nicht intendierten Genomveränderungen unerlässlich. Durch die Anwendung neuartiger Vektorsysteme, wie der AAV9, wird eine effiziente und zielgerichtete Transduktion von Nervenzellen erreicht. Die Möglichkeit der frühen postnatalen Behandlung bietet ein therapeutisches Fenster, um den Progress der Krankheit vor dem Überschreiten toxischer Schwellenwerte zu bremsen oder gar zu verhindern. Die Erforschung des genomischen Verhaltens der Interruptions ermöglicht neue Einblicke in die molekularen Mechanismen der Huntington-Krankheit. So korreliert die Länge der unterbrochenen Repeat-Sequenzen mit der Krankheitsausprägung, was wiederum mit der Rate somatischer Expansionen zusammenhängt.
Die gezielte Veränderung der Sequenzstruktur durch Base-Editing stellt somit eine innovative Manipulation des Krankheitsverlaufs dar. Die Integration von Base-Editing in die klinische Forschung erfordert jedoch noch die Überwindung verschiedener Herausforderungen. Dazu zählen Optimierungen der Auslieferungstechnologien, Minimierung von Nebenwirkungen, Sicherstellung der Stabilität der Korrekturen über den gesamten Lebenszyklus der Zellen sowie die Evaluation der therapeutischen Wirkung bei bereits manifesten Patientenstadien. Besonders faszinierend ist das Potenzial, mit gezieltem Editing präventiv in frühen Lebensphasen oder sogar pränatal einzuwirken, um die pathogene Somatische Expansion zu unterbinden, bevor sie neurodegenerative Prozesse in Gang setzt. Dies könnte zukünftig die Lebensqualität und Prognose von Huntington-Patienten radikal verbessern.
Parallel zu den experimentellen Studien unterstreichen bioinformatische Analysen die Notwendigkeit, die Besonderheiten der mehrmaligen Bindestellen in den Wiederholungstrakten für das Design von sgRNAs zu berücksichtigen, um Präzision und Effizienz zu maximieren. Insgesamt bietet die Anwendung von Cytosin- und Adenin-Base-Editoren eine vielversprechende therapeutische Innovation, die speziell auf die Mutationseigenschaften der Huntington-Krankheit abgestimmt ist. Die Möglichkeit, durch die Einführung natürlicher Interruptions die somatische Repeat-Expansion zu reduzieren, öffnet neue Horizonte in der Behandlung nicht nur von HD, sondern einer Vielzahl polyglutaminer und anderer trinukleotidbasierter Erkrankungen. Die Fortschritte in der Gen-Editing-Technologie gekoppelt mit umfassenden genetischen und molekularen Kenntnissen der Somatischen Repeat-Expansionen könnten den Grundstein für zukünftige klinische Interventionen legen, die den Verlauf der Huntington-Krankheit verzögern oder gar stoppen. Diese multidisziplinäre Herangehensweise vereint molekulare Biologie, Genetik, Neurologie und klinische Medizin und verdeutlicht den Paradigmenwechsel in der Behandlung genetischer Erkrankungen mit bislang unheilbarem Verlauf.
Mit weiteren Studien zur Sicherheit, Langzeitwirkung und therapeutischen Wirksamkeit dieser Methode wird die Hoffnung bestärkt, dass gezieltes Editing von trinukleotidischen Repeats in naher Zukunft klinische Realität wird und somit einen Meilenstein im Kampf gegen neurodegenerative Erkrankungen markieren kann.