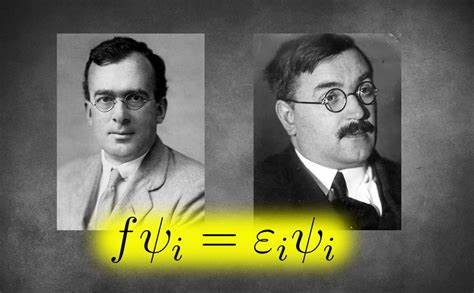
Die Quantenchemie ist ein faszinierendes Gebiet, das die fundamentalen Prinzipien der Chemie und Physik miteinander verbindet. Die Beschreibung der elektronischen Struktur von Molekülen ist dabei eine der zentralen Herausforderungen, denn sie erlaubt es, chemische Eigenschaften und Reaktionen auf atomarer Ebene vorherzusagen. Ein bedeutendes Werkzeug in diesem Kontext ist das Hartree-Fock-Verfahren, eine Methode, die den komplizierten quantenmechanischen Aufbau vieler Elektronensysteme approximativ beschreibt und so die Berechnung energiespezifischer Größen ermöglicht. Helonium, das einfachste Dischtemolekül mit zwei Elektronen, bietet sich als ideales Studienobjekt an, um das Hartree-Fock-Verfahren zu verstehen und anzuwenden. Helonium entstand zu den frühesten Zeiten unseres Universums aus den ersten Wechselwirkungen zwischen Helium und Wasserstoff.
Trotz seiner relativ simplen Struktur birgt das Molekül jedoch eine Vielzahl von komplexen quantenchemischen Eigenschaften, die es zu einem bevorzugten Studienobjekt in theoretischen und rechnerischen Chemieprogrammen machen. Seine zwei Elektronen erlauben es, den Hartree-Fock-Algorithmus in kompakter Form umzusetzen und dennoch eine hohe Aussagekraft zu erlangen. Das Hartree-Fock-Programm, inspiriert von der klassischen und weit verbreiteten Textquelle von Szabo und Ostlund, setzt genau hier an. Es nutzt Basisfunktionen, sogenannte Basis-Sets, um die komplizierten Differentialgleichungen der Quantenmechanik in handhabbare lineare Algebra-Probleme zu überführen. Dabei steht insbesondere die Verwendung von Approximationen im Vordergrund, welche die grundlegenden physikalischen Eigenschaften eines Moleküls mit vertretbarem Rechenaufwand realitätsnah beschreiben.
Ein wichtiger Bestandteil des Programms ist die Verwendung von Slater-Typ-Orbitalen (STO), die die Elektronendichte gut modellieren. Allerdings gestaltet sich die Berechnung der notwendigen elektronischen Integrale mit STOs als sehr anspruchsvoll. Die Lösung hierfür liegt in der Annäherung durch sogenannte Gaussian Type Orbitals (GTO), genauer gesagt im STO-3G Ansatz, der ein STO durch eine gewichtete Summe aus drei Gauß-Funktionen darstellt. Diese Approximation erleichtert die analytische Berechnung der Integrale erheblich, was zu einer dramatischen Verbesserung der Rechenzeiten führt, ohne dabei die physikalische Aussagekraft stark einzuschränken. Die Berechnung der elektronischen Integrale selbst stellt den Kern des Hartree-Fock-Verfahrens dar.
Sie umfassen verschiedene Komponenten wie die Überlappungsintegrale (S), kinetische Energieintegrale (T), Kern-Elektronen-Wechselwirkungsintegrale (V) oder die Zwei-Elektronen-Integrale (ERI). Während einige dieser Integrale analytisch exakt berechnet werden können, bedürfen andere komplexerer numerischer Verfahren oder Approximationen. Dabei spiegeln diese Matrizen letztlich das Verhalten der Elektronen unter dem Einfluss der Atomkerne und der gegenseitigen Elektronenwechselwirkung wider. Die Fock-Matrix ist das zentrale Element, das in jedem Selbstkonsistenten Feld (SCF) Verfahren zum Einsatz kommt. Sie ist eine Näherung zum wahren Hamiltonoperator des Systems, der für die Elektronendynamik verantwortlich ist.
Dieser wichtige Operator wird iterativ optimiert und schließlich so angepasst, dass eine energetisch optimale Lösung gefunden wird. Der iterative Charakter des Verfahrens sorgt dafür, dass anhand der berechneten Dichtematrix systematisch eine verbesserte Fock-Matrix erzeugt wird, bis die Änderung zwischen zwei aufeinanderfolgenden Schritten unter einem vorgegebenen Schwellenwert liegt. Das Hartree-Fock-Programm nutzt außerdem fortschrittliche mathematische Hilfsmittel. Dazu gehört zum Beispiel die Löwdin’sche kanonische Orthogonalisierung, die zur Behandlung der Basisfunktionsüberlappungen eingesetzt wird. Ebenso benötigt das Programm spezialisierte Funktionen zur Berechnung der sogenannten Boys-Funktion, welche bei der Auswertung der Zwei-Elektronen-Integrale eine wesentliche Rolle spielt.
Diese mathematischen Feinheiten gewährleisten, dass die numerischen Approximationen konsistent und physikalisch aussagekräftig bleiben. Ein bemerkenswerter Aspekt des beschriebenen Programms ist dessen Implementierung in der Programmiersprache BQN. Diese ermöglicht eine äußerst kompakte und zugleich effiziente Darstellung der Algorithmen. Mit nur etwa 45 Zeilen Quellcode erreicht die BQN-Variante eine Leistung, die mit der des umfangreichen ursprünglichen Fortran-Programms mit über 500 Zeilen vergleichbar ist. Diese Umsetzung macht das Verfahren nicht nur zugänglicher für Lehrzwecke und Verständnis, sondern auch attraktiv für schnelle praktische Anwendungen und experimentelle Erweiterungen.
Die Selbstkonsistente Feld Methode (englisch Self-Consistent Field, SCF) bildet das Herzstück der Berechnung. Sie ist darauf ausgelegt, ein Näherung des Elektronenzustands schrittweise zu verbessern, indem sie eigenwertähnliche Gleichungen löst, die die Elektronendichte und die Fock-Matrix verknüpfen. Diese Schleife führt letztendlich zu einem stabilen, selbstkonsistenten Zustand, der eine lokale Energie-Minimierung darstellt. Das Verfahren ist stark mathematisch fundiert, beruht auf der Optimierung der Energieerwartungswerte unter Berücksichtigung der Normierungsbedingungen der Wellenfunktion. Die Präzision der Berechnungen mit diesem Programm ermöglicht die Erzeugung sogenannter potenzieller Energieoberflächen, die eine zentrale Rolle in der Beschreibung von Molekülzuständen und chemischen Reaktionen spielen.
Hiermit lassen sich Bindungslängen, Bindungsenergien sowie Übergangszustände von Molekülen theoretisch ermitteln, was sowohl für Grundlagenforschung als auch für zahlreiche Anwendungen in der Materialwissenschaft und der pharmazeutischen Chemie von Bedeutung ist. Helonium als einfaches, aber nicht trivial lösbares System dient gleichzeitig als Evaluationsstandard für Quantenchemie-Software. Die Rechenzeiten und die erzielte Genauigkeit sind entscheidende Kriterien für die Beurteilung unterschiedlicher Implementierungen und Algorithmen. Die Umsetzung in BQN zeigt, dass moderne, funktionale Programmiersprachen mit kompakter Syntax und leistungsfähigen Abstraktionstechniken auch hochkomplexe physikalische Simulationen effizient realisieren können. Neben der unmittelbaren Anwendung auf Helonium ist das vorgestellte Programm so gestaltet, dass es flexibel auf andere Zweielektron-Diatome übertragen werden kann.
Die Struktur des Codes ermöglicht leichtes Anpassen von Potentialparametern und Basis-Sets, wodurch unterschiedliche Molekülsysteme mit vergleichbarem Aufwand analysiert werden können. Diese Eigenschaft macht den Code zu einem hervorragenden Ausgangspunkt für weiterführende Studien und praktischen Einsatz in der Quantenchemie. Der Erfolg des Hartree-Fock-Verfahrens in Kombination mit effizienten numerischen Methoden verdeutlicht, wie weitreichend das Zusammenspiel von theoretischer Physik, angewandter Mathematik und informativer Technik ist. Die Entwicklung solcher Programme zeigt auf beeindruckende Weise, wie klassische Probleme im Bereich der Quantenmechanik von heute modernen algorithmenbasierten Lösungen profitieren. Die kontinuierlichen Verbesserungen in Rechenleistung und Algorithmen eröffnen neue Perspektiven für das Verständnis und die Vorhersage chemischer Systeme.
Zusammenfassend lässt sich sagen, dass das Hartree-Fock Programm für Helonium nicht nur eine akademische Implementierung darstellt, sondern einen wichtigen Baustein in der Ausbildung, Forschung und technologischen Anwendung darstellt. Es vermittelt grundlegende Prinzipien der Quantenchemie, bietet praxisnahe Tools für Molekülsimulationen und zeigt die vielversprechenden Möglichkeiten kompakter Programmierung mit modernen Sprachen wie BQN. So trägt Helonium als molekularer Prototyp dazu bei, das Verständnis der chemischen Bindung und Elektronenkorrelationen auf ein neues Niveau zu heben und den Weg für zukünftige Entwicklungen in der Molekülphysik und Chemie zu ebnen.