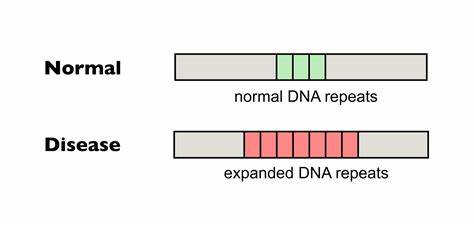
Die Huntington-Krankheit (HD) ist eine verheerende neurodegenerative Erkrankung, die durch eine pathologische Expansion von CAG-Trinukleotid-Wiederholungen im Huntingtin-Gen verursacht wird. Diese Expansionen führen zu einer abnormalen Verlängerung der Polyglutamin (Poly-Q)-Sequenz im Huntingtin-Protein, was schlussendlich zu neuronalen Schäden und dem charakteristischen Abbau motorischer und kognitiver Fähigkeiten führt. Trotz intensiver Forschung existieren bislang keine Heilungsmöglichkeiten, die das Fortschreiten der Erkrankung entscheidend aufhalten können. Aktuelle Entwicklungen in der Genom-Editierung, insbesondere die Präzisionswerkzeuge der sogenannten Base Editing-Technologie, eröffnen jedoch neue Perspektiven in der Bekämpfung dieser Krankheit. Im Zentrum der Huntington-Pathogenese steht die Instabilität der CAG-Wiederholungen im Genom, die sich somatisch ausdehnen können.
Diese somatischen Expansionen tragen wesentlich zum Fortschreiten der Krankheit bei. Untersuchungen zeigten, dass die Länge der reinen, ununterbrochenen CAG-Trakte vergleichsweise besser den Krankheitsverlauf vorhersagt als die Gesamtlänge der Wiederholung. Natürliche Unterbrechungen, beispielsweise durch CAA-Codons innerhalb des CAG-Trakts, sind mit einer höheren Stabilität der Repeats und einer verzögerten oder ausgeprägteren milderen Krankheitsausprägung assoziiert. Solche Unterbrechungen wirken wie Stabilitätsanker und hindern die Bildung schädlicher DNA-Strukturen, die für die Repeat-Expansion verantwortlich sind. Die präzise Einführung solcher Unterbrechungen in pathologische CAG-Trakte ist nun mittels Base Editing realisierbar.
Im Unterschied zu klassischen CRISPR/Cas9-Systemen erzeugt Base Editing keine doppelsträngigen DNA-Brüche, sondern wandelt gezielt einzelne Basen um. Für die Huntington-Krankheit bedeutet dies die gezielte Umwandlung von CAG zu CAA im Huntingtin-Gen, was nicht die Proteinsequenz, sondern lediglich die DNA- und mRNA-Sequenz verändert – ein sogenannter „synonymer“ Austausch. Erste Studien an patientenabgeleiteten Fibroblasten zeigten, dass mit dieser Methode bis zu 80 Prozent der Pathogenese-assoziierten CAG-Repeats mit CAA-Unterbrechungen versehen werden konnten. Diese Bearbeitung führte im Zellkulturmodell zu einer bemerkenswerten Hemmung von weiteren somatischen Expandierungen und teilweise sogar zu einer leichten Kontraktion der Repeatlänge. Methodisch wird das Base Editing durch eine Kombination aus cytosin- oder adenin-Deaminasen mit verbessertem Cas9-Nickase erreicht, die zielgerichtet bestimmte Nukleotide verändern, ohne die DNA-Struktur zu zerstören.
Bei Huntington fokussieren sich die Bearbeitungen auf die Umwandlung von C zu T bzw. G zu A in der DNA, wodurch Interruptionscodons entstehen. Dabei ermöglichen innovative Varianten der Cas9-Nickase, wie Cas9-NG und weitere PAM-erweiterte Varianten, die Ansprache des wiederholten CTG-/CAG-Traktes, der in vielen genomischen Kontexten vorkommt. Die Effizienz und Spezifität dieser Editierungen wurden durch hochauflösende Sequenzierungsverfahren, darunter Hochdurchsatzsequenzierung und Whole-Genome-Sequencing, umfassend überprüft. Auch wurden potenzielle Off-Target-Effekte untersucht.
Die Ergebnisse zeigten einerseits eine hohe Zielgenauigkeit bei der Editierung der CAG-Tracts im HTT-Gen, andererseits die meiste Off-Target-Aktivität im nicht-codierenden oder synonymen Bereich des Genoms. Solche Veränderungen sind häufig nicht schädlich und entsprechen teilweise natürlichen genetischen Varianten. Die Forschung geht über Zellmodelle hinaus und beinhaltet auch Tiermodelle der Huntington-Krankheit, beispielsweise die Htt.Q111-Maus. Durch Adeno-assoziierte Virus (AAV)-vermittelte Abgabe der Base-Editoren in das Neonatalhirn dieser Tiere konnte eine deutliche Reduktion der somatischen Expansionen in relevanten Hirnregionen beobachtet werden.
Diese Intervention führte zur Einführung stabilisierender Interruptionscodons in einem erheblichen Anteil der Huntingtin-Allele und wurde langfristig in Zellen der Großhirnrinde sowie des Striatums nachgewiesen. Wichtigerweise führte dies nicht nur zu einer Hemmung weiterer Expansionen, sondern auch zu einer Rückbildung der Repeatlänge. Die Bedeutung dieser Fortschritte liegt in der potenziellen Verlangsamung des Erkrankungsfortschritts oder sogar deren Verhinderung. Da gemäß neuester molekularer Studien die Expansion der Repeatlänge über Jahrzehnte vor der klinischen Manifestation abläuft, eröffnet die frühzeitige Basis-Editor-Therapie die Chance, diesen Prozess zu unterbrechen. Eine frühzeitige Behandlung könnte dem Patienten so Jahre an gesunder Lebenszeit und Funktionalität schenken.
Neben den präklinischen Daten unterstreichen epidemiologische Untersuchungen die große Bedeutung sogenannter CAA-Unterbrechungen. Populationen mit natürlichen Interruptionscodons weisen signifikant späteres Krankheitsmanifestationsalter auf. Genetische Studien, etwa Genome-Wide Association Studies (GWAS), bestätigen, dass allein das Vorhandensein einer CAA-Unterbrechung das Erkrankungsalter im Durchschnitt um mehr als ein Jahrzehnt verzögert. Neben Huntington könnte das Konzept der Repeat-Interruptions auch für andere trinukleotid-repeat-bedingte Erkrankungen von Bedeutung sein. Friedreich-Ataxie, verursacht durch Expansions von GAA-Repeat-Strecken im FXN-Gen, profitiert ebenfalls von geeigneten Base Editing-Strategien, die Interruptions-Basen im Repeat-Trakt etablieren und so die somatische Expansion verringern.
Die Anwendung von AAV-gestützter Genom-Editierung eröffnete ebenfalls die Diskussion über Langzeitwirkung und Sicherheit. Der dauerhafte Ausdruck von Base-Editoren kann das Risiko unerwünschter Effekte erhöhen, weshalb fortlaufend an weiterentwickelten Deaminase-Varianten gearbeitet wird, die höhere Spezifität besitzen und Nebenwirkungen reduzieren. Auch alternative, temporär wirkende Verabreichungsformen wie virale Virus-like Particles oder mRNA-Delivery sind Gegenstand aktueller Forschungen. Zusammenfassend markieren die jüngsten Fortschritte in der gezielten Bearbeitung von CAG-Repeats mittels Base Editing einen entscheidenden Meilenstein im Kampf gegen Huntington. Die Verringerung somatischer Expansionen in Patientenpräparaten und Tiermodellen zeigt, dass genomische Interruptionscodons direkt eingebaut werden können, um die genetische Stabilität der Repeats zu erhöhen und so den Krankheitsverlauf zu beeinflussen.
Diese Entwicklungen bilden die Grundlage für künftige klinische Studien und die Etablierung sicherer, wirksamer Gentherapien. Es bedarf weiterführender Untersuchungen bezüglich der optimalen Verabreichungswege, der langfristigen Wirkung sowie der Evaluierung möglicher Nebenwirkungen. Die Kombination von molekularer Präzision, immunologischer Verträglichkeit und nachhaltiger Genregulation könnte der Schlüssel zur Behandlung bislang unheilbarer TNR-Erkrankungen wie Huntington sein. In Zukunft könnten vergleichbare Ansätze auch bei familiären Ataxien, bestimmten Spinocerebellar-Ataxien und anderen Polyglutamin-Erkrankungen erfolgreich sein. Grundlegendes Wissen über die Rolle von Menschen-spezifischen Interruptionscodons und deren Mechanismen wird dabei weiterhin essenziell bleiben.
Die Integration von Fortschritten in der Bioinformatik, Hochdurchsatzsequenzierung und personalisierten Medizin ergänzt diese vielversprechenden genetischen Therapiestrategien – ein bedeutender Schritt auf dem Weg zu wirksamen Therapien bei Huntington und verwandten Erkrankungen.