Die computergestützte Chemie hat in den letzten Jahrzehnten eine zentrale Rolle bei der Erforschung von Molekülen und Materialien eingenommen. Sie erlaubt Wissenschaftlern, atomare und molekulare Wechselwirkungen zu simulieren, um Eigenschaften von Substanzen vorherzusagen und neue Materialien zu entwickeln. Ein fundamentaler Baustein dieser Disziplin ist die Dichtefunktionaltheorie (DFT), ein Verfahren, das die komplexen Probleme der Quantenmechanik handhabbar macht. Trotz ihrer weiten Verbreitung und der enormen Bedeutung leidet die DFT jedoch seit langem unter einem zentralen Problem: der begrenzten Genauigkeit der sogenannten Austausch-Korrelations-Funktionale (XC-Funktionale). Diese Funktionale sind entscheidend für die Beschreibung der Elektronendichte und somit für die präzise Berechnung chemischer Eigenschaften.
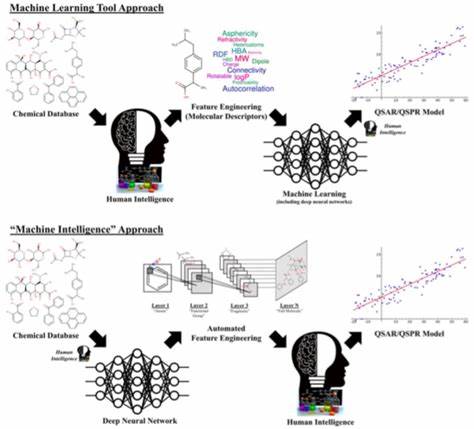
Die traditionelle Formulierung der XC-Funktionale bedient sich dabei meist handgefertigter Modelle, welche in ihrer Fehlergröße häufig das sogenannte chemische Genauigkeitsniveau von etwa 1 kcal/mol deutlich überschreiten. Unsicherheiten in diesem Bereich machen verlässliche Vorhersagen häufig unmöglich und zwingen Forscher, Erkenntnisse oftmals erst durch aufwändige Experimente zu validieren. Mit dem Aufstieg von Künstlicher Intelligenz und insbesondere Deep Learning eröffnen sich nun neue, vielversprechende Wege, um diese begrenzende Hürde zu überwinden. In der jüngsten Forschung wurde eine neuartige Methode entwickelt, welche die Genauigkeit der DFT maßgeblich verbessern kann, indem Deep-Learning-Techniken direkt auf die Modellierung der Austausch-Korrelations-Funktionale angewendet werden. Anstatt sich auf vorab designte, menschlich definierte Merkmale der Elektronendichte zu stützen – sogenannte „Jacob’s Ladder“-Ansätze, die verschiedene Komplexitätsstufen elektrondichtenbasierter Beschreibungen nutzen – lernt das Deep-Learning-Modell eigenständig relevante Repräsentationen aus umfangreichen und äußerst präzisen Daten.
Dieses Prinzip verändert grundlegend die Herangehensweise und ermöglicht sowohl eine Skalierbarkeit als auch eine Genauigkeitssteigerung, die bisher nicht erreichbar war. Ein Kernproblem beim Einsatz von Deep Learning in der DFT lag bislang in der Verfügbarkeit von hochqualitativen Trainingsdaten. Diese Daten stammen aus besonders aufwendigen und rechenintensiven Quantenmechanik-Berechnungen, sogenannten Wellenfunktionmethoden, welche weit genauer als herkömmliche DFT-Ergebnisse sind, aber nur für sehr kleine Molekülsysteme praktikabel bleiben. Um diesem Problem zu begegnen, wurde ein umfangreiches, bisher unerreicht großes Dataset mit hochpräzisen Atomisierungsenergien kleiner Moleküle erstellt. Hierfür wurden Millionen von Rechenstunden auf den leistungsstarken Azure-Computing-Infrastrukturen von Microsoft durchgeführt – ein Aufwand, der sich langfristig durch die Fähigkeit des Modells, diese präzisen Berechnungen auf wesentlich komplexere Systeme zu übertragen, mehr als auszahlt.
Das daraus resultierende Deep-Learning-basierte XC-Funktional, in der Forschung „Skala“ genannt, zeichnet sich durch eine Kombination innovativer Architekturentscheidungen aus, die auf ein effizientes Lernen von nichtlokalen elektronischen Effekten abzielen. Skala erreicht auf dem bekannten W4-17 Benchmark-Datensatz, der Hauptgruppenmoleküle abdeckt, erstmals eine Genauigkeit nahe der chemischen Toleranz von 1 kcal/mol. Mit einer Fehlergröße von 0,85 kcal/mol in einer zentralen Untergruppe von Einzelreferenzsystemen setzt das Modell einen neuen Meilenstein. Dies ist ein Durchbruch, der es erlaubt, DFT-Berechnungen nicht nur zur Interpretation, sondern auch zur zuverlässigen Vorhersage von Experimenten einzusetzen, was bisher undenkbar war. Doch das Potenzial dieser Innovation beschränkt sich nicht nur auf kleinere Moleküle oder thermochemische Eigenschaften.
Die Forschungsgruppe arbeitet bereits an der Erweiterung des Trainingsdatensatzes, um eine breitere Palette chemischer Szenarien abzudecken, darunter komplexere Moleküle und verschiedene molekulare Eigenschaften. Die Skalierbarkeit des Modells und die Effizienz im Vergleich zu klassischen hybriden Funktionalen ermöglichen den Einsatz bei Systemgrößen, die weit über das heutige Standardmaß hinausgehen. Diese Entwicklung könnte den Weg ebnen für eine Simulation-first-Strategie in der Chemie – ähnlich der virtuellen Entwicklung in der Luftfahrtindustrie, wo aufwendige Prototypentests durch präzise Simulationen ersetzt werden konnten. Die Implikationen für Industrie und Forschung sind enorm. In der Pharmaentwicklung können schnelle, verlässliche Vorhersagen der Molekülbindung und Wirkstoffinteraktion die Time-to-Market von Medikamenten drastisch reduzieren.
Im Bereich Energietechnologien erleichtert die verbesserte Modellierung von Materialien die Entwicklung effizienterer Batterien oder umweltfreundlicherer Katalysatoren. Selbst im Bereich der grünen Chemie – etwa bei der Entwicklung nachhaltiger Düngemittel – kann die neue Genauigkeit dabei helfen, ökologische und ökonomische Vorteile zu vereinen. Die Kooperationen mit externen Forschungseinrichtungen und Industriepartnern zeigen ebenfalls, dass dieser Fortschritt nicht nur eine akademische Errungenschaft bleibt. Unternehmen wie Merck betonen die Relevanz für Digital Chemistry-Anwendungen und die Möglichkeit, Workflows der Wirkstoffforschung und Materialentwicklung durch diese neuen Werkzeuge erheblich zu verbessern. Initiativen wie das DFT Research Early Access Program (DFT REAP) ermöglichen es weiteren Akteuren, frühzeitig Zugang zu dieser Technologie zu erhalten und anwendungsorientierte Innovationen voranzutreiben.
Dabei bleibt offen, wie sich diese Technologie in den kommenden Jahren weiterentwickelt. Die generische, skalierbare Infrastruktur und die offene Veröffentlichung von Funktionsbeschreibungen und Datensätzen legen den Grundstein für weitere Forschungen und Optimierungen. Die Verbindung von Deep Learning und wissenschaftlicher Expertise ist dabei von zentraler Bedeutung – das Modell lernt aus großen Datenmengen, wird jedoch durch physikalisch fundierte Einschränkungen, sogenannte Constraints, geleitet, die eine stabile und verlässliche Basis garantieren. Zusammenfassend steht die computergestützte Chemie an der Schwelle zu einer neuen Ära. Die Verbindung von modernsten Deep-Learning-Methoden mit jahrzehntelanger physikalisch-chemischer Forschung überwinden oude Grenzen und eröffnen die Möglichkeit, molekulare Prozesse so präzise und effizient zu simulieren wie nie zuvor.
Dies verspricht eine tiefgreifende Transformation von Forschung und Industrie, bei der computergestützte Vorhersagen Experimente ergänzen oder sogar ersetzen können – mit weitreichenden Folgen für Gesundheit, Energie und Nachhaltigkeit.