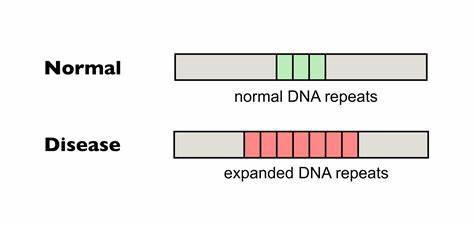
Die Huntington-Krankheit (HD) ist eine verheerende neurodegenerative Erkrankung, die durch die Expansion einer spezifischen trinukleotidischen Wiederholung (CAG-Repeat) im Huntingtin-Gen (HTT) verursacht wird. Diese Expansion führt zu einer verlängerten Polyglutamin-Sequenz im Huntingtin-Protein, die eine toxische Wirkung auf die Nervenzellen ausübt und letztlich zum fortschreitenden Abbau von motorischen und kognitiven Funktionen führt. Trotz zahlreicher Forschungsbemühungen gibt es derzeit keine Heilung für die Huntington-Krankheit, und therapeutische Ansätze zielen vor allem darauf ab, Symptome zu lindern oder den Verlauf zu verlangsamen. In diesem Kontext rückt die präzise Genom-Editierung zunehmend in den Fokus als innovative Strategie, um die genetische Ursache direkt anzusprechen und möglicherweise zu korrigieren. Ein Hauptproblem bei der Huntington-Krankheit ist die Instabilität der CAG-Repeats im Genom, die sich im Laufe des Lebens in somatischen Zellen weiter ausdehnen können.
Diese somatische Expansion trägt entscheidend zum Fortschreiten und zur Schwere der Erkrankung bei. Je länger der Repeat-Abschnitt wird, desto gravierender ist die Zelltoxizität. Das gezielte Eindämmen oder sogar die Umkehrung dieser zusätzlichen somatischen Expansionen ist daher ein vielversprechendes therapeutisches Ziel. Moderne Methoden der Basen-Editierung, insbesondere die sogenannten cytosine und adenine Base Editors (CBE und ABE), ermöglichen es, gezielt einzelne Nukleotide im Genom ohne Doppelstrangbrüche zu verändern. Diese Technik wurde kürzlich erfolgreich angewandt, um die pathogenen CAG-Repeats bei HD und die GAA-Repeats bei Friedreich-Ataxie durch gezielte Einfügung von Unterbrechungen in der Repeat-Sequenz zu modifizieren.
Solche Unterbrechungen kommen natürlicherweise bei einigen gesunden oder weniger stark betroffenen Individuen vor und sind mit stabileren Repeat-Sequenzen sowie verzögerter Erkrankungssymptomatik assoziiert. Im Labor wurde gezeigt, dass das Einfügen von CAA-Codons innerhalb der CAG-Repeats durch Basen-Editierung in Patientenzellen zu signifikant reduzierten somatischen Expansionen führt. Die so modifizierten Sequenzen bilden stabilere Strukturen und sind weniger anfällig für fehlerhafte DNA-Reparaturmechanismen, die für die Expansion verantwortlich sind. Diese durch Basen-Editierung erzeugten Unterbrechungen sind synonym, das heißt, sie verändern nicht die Aminosäuresequenz des resultierenden Huntingtin-Proteins und vermeiden somit direkte Proteinmodifikationen, die unerwünschte Effekte hervorrufen könnten. Ebenso konnten Studien an Huntington-Mausmodellen demonstrieren, dass die Anwendung von viralen Vektoren für die In-vivo-Übertragung von Base Editors in das zentrale Nervensystem zu einer nachhaltigen Einführung von CAA-Unterbrechungen am HTT-Gen führt.
Die Folge war eine signifikante Verringerung der somatischen CAG-Repeat-Expansion in Gehirnregionen, die besonders von HD betroffen sind, wie etwa Kortex und Striatum. Interessanterweise kam es dabei nicht nur zu einer Stabilisierung der Repeat-Länge, sondern vereinzelt auch zu einer Kontraktion der Repeats, also einer Verkürzung der pathogenen Sektion. Neben huntingtinbezogenen Repeats sind ähnliche Prinzipien auch bei anderen trinukleotidaren Repeat-Erkrankungen, wie der Friedreich-Ataxie, anwendbar. Dort ist die Expansion von GAA-Repeats im FXN-Gen verantwortlich für die Erkrankung. Adenin-Base-Editing konnte in Patientenzellen und in Tiermodellen GAA-Unterbrechungen erzeugen, die mit einer verbesserten FXN-Genexpression und einer Reduktion somatischer Repeat-Expansionen einhergehen.
Diese Ergebnisse verdeutlichen die Übertragbarkeit und das Potenzial der Basen-Editierung als universelle Methode zur Behandlung einer Vielzahl von Repeat-Expansionserkrankungen. Ein entscheidender Vorteil der Basen-Editierung gegenüber klassischen CRISPR/Cas9-Techniken liegt im Verzicht auf Doppelstrangbrüche im DNA-Strang, welche häufig mit unerwünschten Nebenwirkungen wie Indels oder chromosomaler Instabilität einhergehen. Dies ist besonders wichtig bei der Therapie von Erkrankungen, bei denen die Genomintegrität in postmitotischen Zellen des Nervensystems gewährleistet werden muss. Sicherheitsbedenken bezüglich off-target Effekten bei der Basen-Editierung konnten durch differenzierte Untersuchungen weitestgehend entspannt werden. Genome-weite Analysen zeigten, dass die meisten unbeabsichtigten Basenänderungen entweder in nicht-kodierenden Regionen des Genoms auftreten oder zu synonymen Codierungsänderungen führen, die keine funktionellen Auswirkungen auf Proteine haben.
Lediglich ein geringer Anteil von Off-target-Editierungen betrifft proteinkodierende Regionen, wobei viele dieser Veränderungen als benign bewertet werden konnten. Dennoch bleibt der sorgfältige Umgang mit potenziellen Nebeneffekten wichtig, insbesondere für die zukünftige klinische Anwendung. Die Effizienz der Basen-Editierung wurde zudem durch die Wahl geeigneter zielgerichteter Adenin- und Cytosin-Deaminasen sowie optimierter Cas9-Varianten mit erweiterten PAM-Erkennungssequenzen deutlich verbessert. Dies ermöglicht die gezielte Bearbeitung selbst langer und repetitiver Abschnitte, die zuvor als schwierig galten. Die Verabreichung der Basis-Editoren erfolgt häufig über Adeno-assoziierte Viren (AAVs), deren Tropismus auf das zentrale Nervensystem spezialisiert ist.
Neonatale Injektionen in Modelltieren führten zu hoher Transduktion in Gehirnregionen und einer nachhaltigen Basen-Editierung über Monate hinweg. Optimierungen hinsichtlich Dosierung, Vectordesigns und Verabreichungsrouten werden derzeit für eine bessere Krankheitsrelevanz weiter erforscht. Das Konzept, somatische Repeat-Expansionen frühzeitig im Krankheitsverlauf durch Basen-Editierung zu verhindern bzw. zu reduzieren, könnte theoretisch das Fortschreiten neurologischer Symptome verzögern oder sogar verhindern. Aktuelle Modelle deuten darauf hin, dass HD-Neuronen lange Zeit mit zunehmender CAG-Repeat-Länge leben, bevor eine kritische Schwelle überschritten wird, die zu rascher Neurodegeneration führt.
Durch den Einsatz von Basen-Editoren könnte diese Schwelle möglicherweise erst gar nicht erreicht werden. Zusammenfassend steht die gezielte Unterbrechung pathogener trinukleotidärer Repeat-Sequenzen mittels Basen-Editierung für einen innovativen Ansatz in der Behandlung der Huntington-Krankheit und verwandter TNR-Erkrankungen. Diese Technologie bietet eine Kombination aus präziser Genomkorrektur, reduzierter Nebenwirksamkeit und nachhaltiger Wirkung in betroffenen Geweben. Obwohl weitere präklinische Studien und Sicherheitsüberprüfungen notwendig sind, eröffnen die bisherigen Erkenntnisse neue Hoffnungsschimmer für Betroffene dieser bisher unheilbaren Erkrankungen. Zukünftige Forschungsarbeiten fokussieren sich auf die Erweiterung der Methodik auf weitere TNR und die Verbesserung der Editoreffizienz sowie der gezielten Verabreichung in unterschiedliche Zelltypen und Organsysteme.
Auch die Entwicklung transienter und kontrollierbarer Expressionseinheiten sowie alternative Lieferformen wie virale Vektoren mit geringerer Immunogenität oder virale Partikel ohne Genintegration (Virus-like Particles) werden erforscht. Die Integration von Basen-Editierung in das therapeutische Repertoire gegen Huntington könnte letztendlich einen Paradigmenwechsel darstellen – weg von symptomatischen Behandlungen hin zu zielgerichteten molekularen Interventionen, die die genetische Ursache direkt und nachhaltig adressieren. Dieses Potenzial macht die präzise Genom-Editierung zu einer der spannendsten Felder in der modernen Neurologie und Genetik. Fortschritte in der Erforschung der somatischen Repeat-Instabilität und deren Modifikatoren ermöglichen zudem eine immer bessere Identifikation von Zielregionen und geeigneten Editoren. Eine Kombination dieser Therapieansätze mit weiteren molekularen und zellulären Therapien, beispielsweise Antisense-Oligonukleotiden oder RNA-Interferenz, könnte in Zukunft synergistische Effekte entfalten und die Lebensqualität der Patienten weiter verbessern.
Abschließend lässt sich sagen, dass die Basen-Editierung von trinukleotidischen Repeats bei der Huntington-Krankheit einen vielversprechenden Schritt hin zu einer dauerhaften und ursächlichen Therapie darstellt. Die stetige Optimierung dieser Technologie und deren erfolgreiche Anwendung in präklinischen Modellen bilden die Grundlage für zukünftige klinische Studien und potenzielle Behandlungsstrategien für Patienten mit Huntington und verwandten neurodegenerativen Erkrankungen.