Die Evolution des Menschen ist eine der faszinierendsten Geschichten der Naturwissenschaften, doch trotz jahrzehntelanger Forschung bleiben zahlreiche Fragen zu unserer Herkunft und der komplexen demographischen Geschichte offen. Ein Meilenstein in der Aufklärung dieser Vergangenheit sind neue methodische Ansätze, die es ermöglichen, genetische Signale uralter Ereignisse aus dem menschlichen Genom zu entschlüsseln. Eine solche innovative Methode ist das strukturierte Coaleszenz-Modell, das jüngst wegweisende Erkenntnisse über die tiefe Ahnenstruktur der modernen Menschen eröffnet hat. Die Coaleszenz-Theorie ist ein zentraler Bestandteil der Populationsgenetik und erlaubt es, die genealogische Geschichte von Genen rückwärts in die Vergangenheit zu rekonstruieren. Klassische Modelle basieren dabei oft auf der Annahme einer panmiktischen Population – das heißt, eine Population, in der alle Individuen zufällig miteinander paaren und die keine Struktur aufweist.
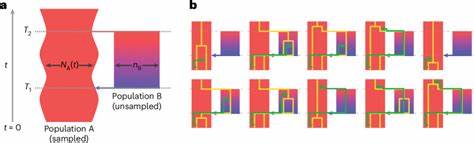
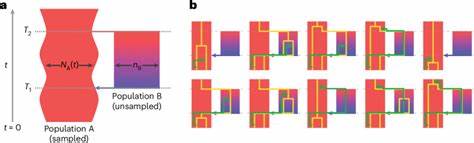
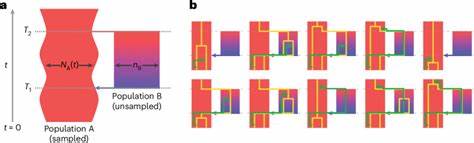
Doch es ist immer deutlicher geworden, dass diese Annahme für die komplexen evolutionären Prozesse bei Homo sapiens nicht ausreichend ist. Die neue Studienarbeit von Trevor Cousins, Aylwyn Scally und Richard Durbin bringt deswegen ein strukturiertes Coaleszenz-Modell namens "cobraa" zum Einsatz, das ancestral divergierende Populationen mit anschließender Rekombination der Gene abbildet. Das Modell berücksichtigt ein Szenario, in dem sich eine Ursprungsbevölkerung vor etwa 1,5 Millionen Jahren in zwei getrennte Gruppen aufteilte, die dann vor rund 300.000 Jahren wieder miteinander verschmolzen sind. Dabei trat eine Mischung der ahnenmäßigen Linien im Verhältnis von etwa 80 zu 20 Prozent auf.
Diese Erkenntnis ist besonders spannend, da sie nahelegt, dass alle heutigen Menschen Spuren dieser tiefen, gemeinsam geteilten Ahnenstruktur in sich tragen und dass die Evolution des modernen Menschen keine einfache, linear verlaufende Geschichte war. Ein zentrales Element des Modells ist die Fähigkeit, durch eine Hidden-Markov-Struktur zwischen den Übergangswahrscheinlichkeiten der Coaleszenzzeiten einzelner Genome zu unterscheiden – und damit strukturierte von unstrukturierten demographischen Szenarien abzugrenzen. Dies ist ein bedeutender Fortschritt, weil frühere Methoden wie das Paarweise Sequenzielle Markowsche Coaleszenz-Modell (PSMC) zwar effektiv Veränderungen in der Populationsgröße erfassen, aber strukturelle Unterteilungen in der Vergangenheit nicht eindeutig detektieren konnten. Verbunden mit diesem Modell sind zudem Hinweise auf eine starke Flaschenhals-Situation in der größeren der beiden frühen Populationen unmittelbar nach deren Aufspaltung. Das Vorhandensein eines solchen Flaschenhalses, also einer dramatischen Reduktion der effektiven Populationsgröße, erklärt einige vorher beobachtete genetische Signaturen und stimmt mit Fossilfunden aus der Frühphase des Homo sapiens überein.
Außerdem fanden die Wissenschaftler heraus, dass die DNA-Abschnitte, die von der kleineren Population stammen, statistisch stärker mit dekodierenden Genabschnitten in der Nähe korrelieren und dabei tendenziell nachteilig sind. Dies lässt vermuten, dass diese genetischen Varianten weniger günstig waren und durch Selektion gegen sie gearbeitet wurde. Eine weitere bedeutende Auswertung zeigte eine starke Korrelation zwischen der genomischen Herkunft aus der größeren der zwei Urbevölkerungen und der Ähnlichkeit zu den Neandertalern und Denisovanern, den bekannten archaischen Verwandten des modernen Menschen. Daraus wird geschlossen, dass diese Hauptlinie auch die gemeinsame Vorfahrenschaft zu den archaischen Homininen bildet und somit indirekt die genetische Verfügbarkeit von Genen für spätere Mischungen mit Neandertalern und Denisovanern bereitstellte. Die Anwendung des Modells auf umfangreiche Datensätze wie das 1000 Genomes Project und das Human Genome Diversity Project bestätigte die Präsenz dieser tiefen Struktur bei nahezu allen untersuchten menschlichen Populationen, einschließlich der Khoisan, die als genetisch besonders vielfältig und divergent gelten.
Dieses Ergebnis stellt eine wesentliche Neuerung gegenüber früheren Modellen dar, die oft keine oder nur sehr junge Strukturereignisse nachwiesen. Beachtenswert ist, dass die vorherrschende Annahme einer durchgängigen panmiktischen Bevölkerung in der Vergangenheit nicht ausreichend die genetischen Daten erklärt. Stattdessen zeigt das strukturierte Modell einen wesentlich komplexeren Verlauf der menschlichen Vorfahren, der unter anderem Phasen der Isolation, Divergenz und erneuten Vermischung umfasst – ein Szenario, das durch archäologische Erkenntnisse und Fossilfunde an mehreren Orten der Welt gestützt wird. Das Modell wurde zudem auf weitere Säugetierarten angewandt, darunter Fledermäuse, Delfine, Gorillas und Schimpansen. Die Ergebnisse variierten, was die methodische Empfindlichkeit und gleichzeitig die biologische Vielfalt der Populationsgeschichten verschiedener Spezies widerspiegelt.
Beispielsweise fand sich bei den parti-colored bats kaum Evidenz für eine solche tiefe Struktur, während bei Delfinen eine vergleichbare Mischung zweier Populationen nachgewiesen wurde. Dies demonstriert die Flexibilität und Breite der Anwendungsmöglichkeiten des neuen Modells. Die Verwendung von Hochdurchsatz-Sequenzierungsdaten ermöglichte dabei eine hochauflösende Analyse der genetischen Variation. Technisch baut das Modell auf der Erweiterung der bereits bewährten Hidden-Markov-Methoden aus, beinhaltet jedoch zusätzliche Zustände, die die Wege der genetischen Abstammung über die beiden angenommenen ursprünglichen Populationen verfolgen. Dies erlaubt sowohl die Modellierung von demographischen Prozessen als auch das Inferenzieren von Genabschnitten mit unterschiedlichem Ursprungsstatus.
Genauere Analysen der genomischen Verteilung der cis- und transintrogressiven Genabschnitte zeigten, dass bestimmte biologische Funktionen überproportional in den mit „minoritärem“ Ursprung assoziierten Regionen vertreten sind. Insbesondere Gene, die mit neuronaler Entwicklung, neuronaler Erkennung und Verbindung, sowie synaptischer Übertragung im Gehirn assoziiert sind, wiesen eine stärkere Überpräsenz in Regionen mit höherem Anteil an der kleinen Urbevölkerung auf. Im Gegensatz dazu waren bestimmte genetische Prozesse wie miRNA-Verarbeitung, Immunantwort oder sensorische Wahrnehmungen in diesen Regionen unterrepräsentiert. Diese Befunde verweisen darauf, dass die urtümliche Vermischung auch funktionale Anpassungen oder Selektionsprozesse widerspiegelt, die für die Evolution der menschlichen Spezies relevant waren. Die Studie bringt damit eine wichtige Komponente in das Verständnis der menschlichen Demographie, die Einfluss auf genealogi-sche Rekonstruktionen, die Interpretation archaischer Genflussereignisse und darauf aufbauende Modelle der Menschheitsgeschichte hat.
Das erkannte Ereignis der tiefen Populationsteilung und das anschließende Wiederzusammenschmelzen waren offenbar ein prägender Moment in der Entstehung der genetischen Vielfalt und Struktur, die wir heute in allen modernen Bevölkerungen vorfinden. Diese moderne Methodik eröffnet aber nicht nur neue Erkenntnisse zur Vergangenheit, sondern bietet auch Perspektiven für die Erforschung von Struktur- und Mixereignissen in anderen Arten und für die Analyse komplexer demographischer Prozesse unter Berücksichtigung von Flaschenhälsen, Selektion und Archaischem Genfluss. Zudem erweitert sie die Werkzeuge der Evolutionsbiologie, indem sie den Informationsgehalt der Verteilung benachbarter genealogischer Ereignisse nutzt, um bisher unerkennbare Ereignisse in der Evolution zu identifizieren. Dennoch sollten die Ergebnisse in ihrem komplexen methodischen und biologischen Kontext betrachtet werden. Das Modell beruht auf Annahmen wie Abwesenheit von Selektion, konstanter Mutations- und Rekombinationsraten, sowie der Einfachheit eines einzigen Pulse-Events der Vermischung.
Die Realität der menschlichen Vergangenheit ist höchstwahrscheinlich noch komplexer, mit multiplen Admixturen, kontinuierlichem Genfluss und heterogenen Selektionsdruckmustern, die weitere methodologische Anpassungen und mehr genomische Daten erfordern. Zusammenfassend zeigt die Anwendung des strukturierten Coaleszenz-Modells, dass die menschliche Evolution durch eine lange Phase der Divergenz zwischen zwei großen Urgruppen und deren spätere Vermischung geprägt wurde. Diese Erkenntnis verbindet genetische, archäologische und fossile Befunde zu einem kohärenten Bild der langanhaltenden Struktur und Dynamik, die alle heutigen Menschen miteinander teilen. Die fortschreitenden technologischen und theoretischen Entwicklungen versprechen in Zukunft noch tiefere Einsichten in die vielschichtige Geschichte des Homo sapiens und anderer Arten.