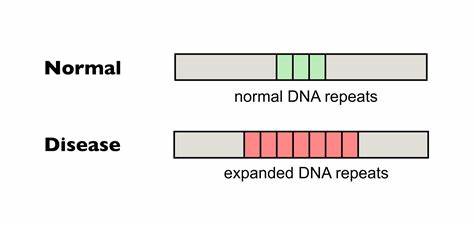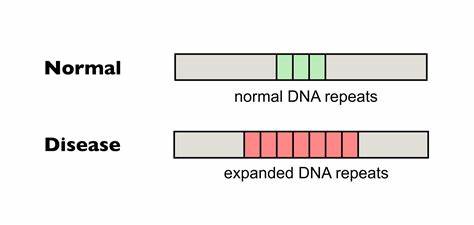
Huntington ist eine komplexe neurodegenerative Erkrankung, die durch die Expansion von trinukleotidischen Wiederholungen im sogenannten HTT-Gen ausgelöst wird. Im Kern der Pathogenese steht eine instabile Sequenz von CAG-Wiederholungen, deren verlängerte Abschnitte Proteinfaltungsstörungen verursachen und schließlich die neuronal-degenerativen Symptome hervorrufen. Die Anzahl der CAG-Wiederholungen ist nicht nur ein wesentlicher Prädiktor für das Erkrankungsalter, sondern auch für die Geschwindigkeit des progressiven Abbaus neuronaler Funktionen. Dabei ist bekannt, dass sich die Repeats innerhalb des Körpers, insbesondere im Zentralnervensystem, im Somagewebe dynamisch weiter verlängern – ein Phänomen, das als somatische Expansion bezeichnet wird und zur Verschlimmerung der Symptome beiträgt. Aufgrund dessen gilt die Reduktion dieser somatischen Expansion als ein vielversprechendes therapeutisches Ziel, um den Krankheitsverlauf aufzuhalten oder zumindest zu verlangsamen.
In den vergangenen Jahren hat die Genom-Editierung erhebliche Fortschritte erlebt, speziell durch die Entwicklung von Basen-Editoren. Diese Werkzeuge ermöglichen präzise Einzelbasenveränderungen in der DNA, ohne die Erzeugung von Doppelstrangbrüchen, die zu unerwünschten Mutationen führen können. Besonders relevant für Huntington ist die Möglichkeit, mittels cytosin- oder adeninbasierter Editoren, sogenannte 'Unterbrechungen' in die CAG-Repeatsequenz einzubauen. Diese Unterbrechungen verändern einzelne Basen innerhalb der Trinukleotidkette, beispielsweise durch die Umwandlung der CAG-Codons in CAA-Codons, welche zwar dieselbe Aminosäure Glutamin kodieren, jedoch die DNA-Sequenz stabilisieren. Solche Interruptionssequenzen kommen natürlich auch bei manchen Menschen vor, die eine mildere Verlaufsform der Krankheit zeigen oder ein verspätetes Erkrankungsalter aufweisen.
Aktuelle Forschungsarbeiten demonstrieren, dass die gezielte Einführung dieser Unterbrechungen durch Basen-Editierung zu einer signifikanten Verringerung der somatischen Expansion in patienteneigenen Fibroblasten sowie in Mausmodellen führt. So wurde gezeigt, dass die Verabreichung von optimierten Cytosin-Basen-Editoren (CBE) gemeinsam mit präzise designten sgRNAs, die spezifisch an die CTG-Sequenzen binden, bis zu 80 % der Zellen Editing an den HTT-Allelen erreichten und damit eine dauerhafte Modifikation erzeugten. Darüber hinaus bleiben diese Veränderungen stabil über mehrere Zellgenerationszyklen, was ihre therapeutische Absicht unterstreicht. Neben den Zellkulturstudien wurden die Editoren in vivo mittels Adeno-assoziierter Virusvektoren mit Serotyp 9 (AAV9) erfolgreich in das zentrale Nervensystem von neonatalen Huntington-Mausmodellen eingebracht. Die Neurospezifität von AAV9 ermöglicht es, die Zielregionen wie Cortex und Striatum zu transduzieren – Areale, die bei Huntington besonders betroffen sind.
In den behandelten Mäusen konnten konsistent eine erhöhte Frequenz von CAA-Unterbrechungen und eine deutlich verminderte somatische Expansion der CAG-Tracts beobachtet werden. Erstaunlicherweise führte die Behandlung nicht nur zum Stillstand der Expansion, sondern förderte sogar eine Verkürzung der Wiederholungen, was einen zusätzlichen therapeutischen Vorteil darstellt. Die molekularen Mechanismen hinter der heilenden Wirkung dieser Unterbrechungen liegen in der beeinträchtigten Bildung stabiler sekundärer Strukturen in der DNA, welche sonst DNA-Reparaturmechanismen irreführen und fehleranfällige Erweiterungen der Repeatlänge verursachen. Unterbrochene Repeatsequenzen verhindern die Entstehung solcher Strukturen, verringern DNA-Schleifenbildung und R-Loops und stabilisieren so den Genomabschnitt. Diese Stabilisierung führt zu einer reduzierten Aktivität macher DNA-Reparaturprozesse, die bei der Expansion wesentlich beteiligt sind.
Trotz der positiven Ergebnisse wirft die Verwendung von Basis-Editoren neue Fragen hinsichtlich der Sicherheit auf. Eine Herausforderung besteht darin, ungewollte Off-Target-Effekte zu minimieren. Systematische Analysen zeigen, dass obwohl CBE und ABE bevorzugt an den Zielstellen wirken, in geringerer Häufigkeit auch andere genomische Loci mit ähnlichen Sequenzmotiven bearbeitet werden können. Die meisten dieser Off-Target-Effekte scheinen sich jedoch auf nicht kodierende oder synonym kodierende Bereiche zu beschränken und verursachen selten schädliche Auswirkungen. Dennoch wird eine umfassende Langzeitüberwachung in Tiermodellen empfohlen, um alle potenziellen Risiken zu evaluieren und Strategien zur Sicherung der klinischen Anwendbarkeit zu entwickeln.
Die grundsätzliche Zugänglichkeit und Flexibilität von Basen-Editoren macht sie zu einer vielversprechenden Plattform, nicht nur für die Behandlung von Huntington, sondern auch für andere trinukleotidbasierende Erkrankungen. So wurden vergleichbare Ansätze auch bei Friedreich-Ataxie mit GAA-Repeat-Expansionen im FXN-Gen erfolgreich getestet und konnten dort ähnlich stabile Unterbrechungen einführen, die zum geringeren somatischen Wachstum der Repeats führten. Die gemeinsame Grundlage dieser TNR-Erkrankungen bestätigt die vielseitige Einsetzbarkeit der Basen-Editor-Technologie. Die nächsten Schritte für die Translation dieser Technologie in klinische Anwendungen beinhalten unter anderem die Optimierung der Liefersysteme, um sicher und effizient in menschliche Gewebe zu gelangen, die Reduktion der Off-Target-Rate durch Fine-Tuning der Enzymkomponenten sowie die Verfeinerung der sgRNA-Designs zur Maximierung der Zielgenauigkeit. Weiterhin sind klinisch relevante Tiermodelle notwendig, die nicht nur molekulare Effekte, sondern auch verbesserte motorische und kognitive Funktionen zeigen.
Noch ein nicht vernachlässigbarer Aspekt ist der ethische Umgang mit genetischen Eingriffen, die eine dauerhafte Veränderung des Genoms mit sich bringen. Zusammenfassend lässt sich festhalten, dass die gezielte Basen-Editierung von CAG-Repeat-Sequenzen im HTT-Gen eine der innovativsten Strategien darstellt, um die somatische Expansion zu unterbinden, welche eine zentrale pathologische Komponente bei Huntington ist. Die Möglichkeit, die Expansion zu stabilisieren und sogar zu reduzieren, eröffnet faszinierende Perspektiven für eine präventive und therapeutisch wirksame Behandlung, die über die symptomatische Linderung hinausgeht. Während weitere Forschung notwendig ist, um die Sicherheit und Effektivität beim Menschen zu gewährleisten, bietet die Basis-Editor-Technologie eine realistische Aussicht auf transformative Neuerungen in der Behandlung von Huntington und verwandten trinukleotidbasierten Erkrankungen.