Huntington ist eine verheerende neurodegenerative Erkrankung, hervorgerufen durch die pathologische Expansion von CAG-Trinukleotidwiederholungen im HTT-Gen. Diese genetischen Wiederholungen führen zur Produktion eines mutierten Huntingtin-Proteins, das für die progressive Degeneration von Nervenzellen verantwortlich ist. Charakteristisch für den Verlauf der Krankheit ist nicht nur die Länge der genetischen Wiederholung bei Geburt, sondern auch ihre weitere somatische Expansion, welche mit zunehmendem Alter die Krankheitsprogression beschleunigt. Bis vor kurzem gab es keinerlei therapeutische Ansätze, die gezielt die somatische Instabilität und Expansion der CAG-Wiederholungen beeinflussen konnten. Dies ändert sich mit dem Aufkommen präziser Genome-Editierungstechnologien, insbesondere dem sogenannten Basen-Editing, das nun vielversprechende Ergebnisse bei der Verringerung von somatischen Repeat-Expansionen in Patienten-Zellen liefert.
Die Instabilität der CAG-Repeats ist ein dynamischer Prozess, der im Verlauf des Lebens weiter voranschreiten kann. Studien zeigen, dass Neuronen von Huntington-Patienten über Jahrzehnte eine Zunahme der CAG-Repeat-Länge erfahren, bevor Symptome auftreten. Dieser Prozess wird als somatische Expansion bezeichnet und gilt als bedeutender Einflussfaktor für das Fortschreiten der Erkrankung. Die somatische Expansion beruht auf komplexen DNA-Reparaturmechanismen, die während der Zellteilung und Transkription die Wiederholungslänge verändern. Dabei begünstigen spezifische DNA-Strukturen und R-Loops die Fehlerraten bei der Reparatur, was zu einer weiteren Verlängerung der Wiederholungen führt.
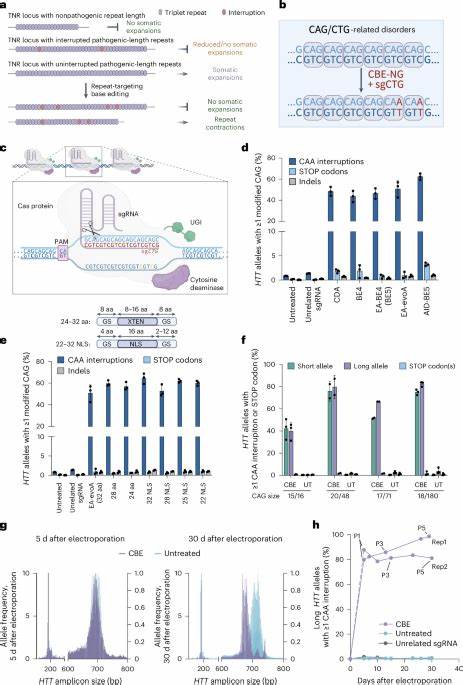
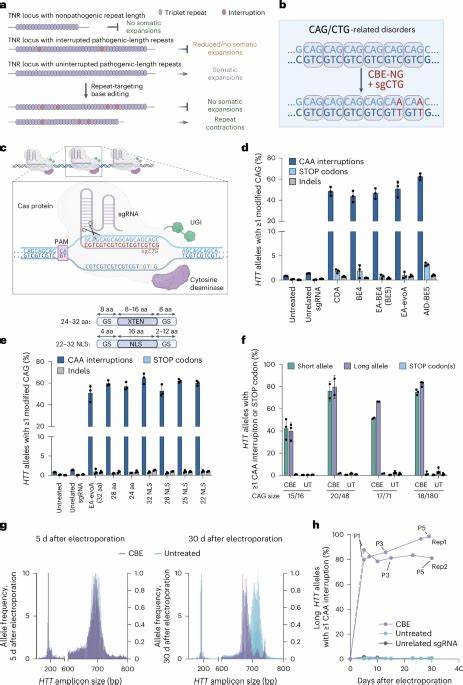
Interessanterweise besitzen manche natürliche Allele kleine Unterbrechungen in der CAG-Trinukleotidkette, die sich als Schutzfaktor gegen somatische Instabilität zeigen. Beispielsweise können CAA-Triplets innerhalb der CAG-Repeats eingebaut sein, ohne dass sie die Aminosäuresequenz des Huntingtin-Proteins verändern. Solche Unterbrechungen sind mit einer Stabilisierung des genetischen Locus und einem verzögerten Krankheitsbeginn assoziiert. Darauf aufbauend entstand die Idee, mit Basen-Editoren gezielt diese Unterbrechungen in das pathogene HTT-Gen einzufügen, um auf molekularer Ebene die Somatik-Instabilität zu reduzieren und so den Krankheitsverlauf positiv zu beeinflussen. Basen-Editing ist eine innovative Form der Genom-Editierung, die ohne Doppelstrangbrüche direkt einzelne Basenpaare an spezifischen DNA-Stellen umwandelt.
Dabei werden Cytosin- oder Adenin-Basen in andere Basen umgewandelt, was präzise und effizient kleine, aber wichtige Veränderungen im Genom bewirkt. Im Fall von Huntington zielen Forscher darauf ab, CAG-Sequenzen durch Einführung von CAA-Unterbrechungen zu verändern, was die sogenannte „Repeat-Purezza“ reduziert und somit die Anfälligkeit für Expansion mindert. Jüngste Studien berichten, dass solche Basen-Editoren in vitro in Fibroblasten von Huntington-Patienten erfolgreich eingesetzt werden können. Nach der Anwendung der Technologie zeigen die Zellen eine signifikante Reduktion der somatischen Expansion der CAG-Wiederholung. Dies bedeutet, dass die genetische Wiederholung durch die eingefügten Unterbrechungen stabiler wird und weniger zu unkontrollierter Verlängerung neigt.
Zudem wurde beobachtet, dass die Bearbeitung des mutierten langen HTT-Allels effektiver ist als des normalen kurzen Allels, was einen therapeutischen Vorteil verspricht. Der Weg von der Zellkultur zum lebenden Organismus gelang durch präklinische Studien an Mausmodellen, die humane Huntington-Allel-Trinukleotid-Längen tragen. Dort wurde gezeigt, dass die Verabreichung von AAV9-Vektoren, die den Basen-Editor und die zielgerichtete sgRNA enthalten, zu einer nachhaltigen und effizienten Einführung der CAA-Unterbrechungen in relevanten Hirngebieten führt. In den behandelten Tieren konnte eine signifikante Verringerung der somatischen Expansion in den Neuronen des Gehirns nachgewiesen werden. Gleichzeitig wurden keine schädlichen Nebenwirkungen oder signifikanten Off-Target-Effekte festgestellt, was die Sicherheit der Methode unterstreicht.
Es stellt sich die Frage, wie breit die Anwendung von Basen-Editing im Kontext von Huntington und anderen ähnlichen trinukleotidbedingten Erkrankungen ist. Die Grundprinzipien der Technologie ermöglichen theoretisch die Anpassung an verschiedene genomische Repeat-Sequenzen. So werden auch bei der Friedreich-Ataxie (einer Erkrankung mit GAA-Repeat-Expansion) ähnliche Unterbrechungsstrategien erforscht. Die erfolgreiche Reduktion somatischer Expansionen in tierischen Modellen lässt auf einen breiten therapeutischen Nutzen schließen. Die Potenziale dieser neuen therapeutischen Ansätze sind enorm.
Durch die dauerhafte Stabilisierung der Trinukleotid-Wiederholungen können die neurodegenerativen Prozesse hinausgezögert oder möglicherweise sogar verhindert werden. Dies würde für Betroffene eine deutliche Verlängerung der symptomfreien Lebensjahre bedeuten und die Lebensqualität verbessern. Gleichzeitig eröffnet das präzise Basen-Editing neue Perspektiven für personalisierte Medizin, da die Therapie spezifisch auf die individuelle Repeat-Länge und -Konstitution angepasst werden kann. Dennoch gibt es Herausforderungen, die es zu bewältigen gilt. Die effiziente und sichere Auslieferung von Basen-Editoren in die relevanten Zelltypen des menschlichen Gehirns bleibt komplex, insbesondere bei erwachsenen Patienten.
Zudem sind langfristige Studien notwendig, um mögliche unerwünschte Editierungen und Immunreaktionen auszuschließen. Nicht zuletzt müssen regulatorische Hürden überwunden werden, um diese neuartige Gentherapie in die klinische Praxis zu integrieren. Fazit ist, dass durch die innovative Nutzung von Basen-Editing ein vielversprechender Ansatz vorhanden ist, die somatische Expansion der CAG-Trinukleotid-Wiederholungen bei Huntington effektiv zu vermindern. Die Einführung natürlicher und harmloser Wiederholungsunterbrechungen in das mutierte HTT-Gen stabilisiert die Repeat-Sequenz und verlangsamt die Krankheitsprogression auf genetischer Ebene. Die bisherigen präklinischen Erfolge bei Patienten-Zellen und Tiermodellen bilden das Fundament für zukünftige klinische Studien, welche die Wirksamkeit und Sicherheit beim Menschen belegen müssen.
Die Fortschritte in der molekularen Gentherapie bieten somit neue Hoffnung für Menschen, die von Huntington und anderen Repeat-Expansion-Erkrankungen betroffen sind.