Die Huntington-Krankheit (HD) gehört zu den sogenannten trinukleotidischen Wiederholungserkrankungen, deren Ursache auf die pathologische Erweiterung bestimmter DNA-Sequenzen beruht. Im Speziellen handelt es sich bei HD um eine Erweiterung von CAG-Trinukleotidrepeats im Huntingtin-Gen (HTT). Diese Erweiterungen führen schließlich zur Bildung toxischer Proteine, die Nervenzellen schädigen und zu den charakteristischen motorischen, kognitiven sowie psychischen Symptomen der Erkrankung führen. Obwohl die genetische Grundlage von HD seit Jahrzehnten bekannt ist, bleiben effektive kurative Therapien bislang aus. Aktuelle Forschungsansätze konzentrieren sich deshalb neben symptomatischen Behandlungen verstärkt auf die molekulare Manipulation der DNA-Sequenzen, die die Krankheit auslösen.
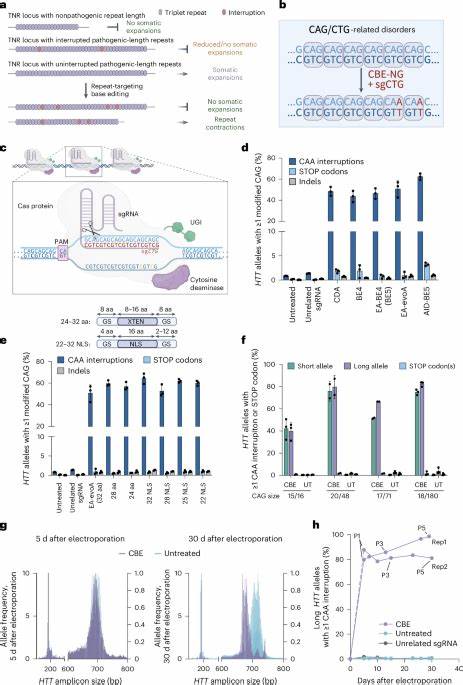
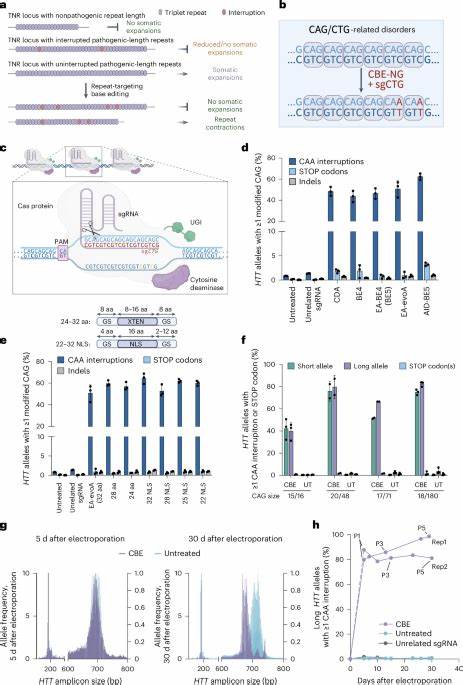
Ein innovativer Strategieansatz besteht darin, die CAG-Repeats direkt im Genom der Patienten zu bearbeiten – mit dem Ziel, die somatische Expansion, also die progressive Vermehrung dieser Wiederholungen in Körperzellen, zu verhindern oder zu reduzieren. Trinukleotidwiederholungen wie die CAG-Sequenzen können instabil sein und sich insbesondere in Nervenzellen im Laufe der Lebenszeit weiter verlängern. Diese somatische Expansion spielt eine entscheidende Rolle für den Krankheitsverlauf, da längere CAG-Abschnitte mit einem früheren Krankheitsbeginn und schnelleren Fortschreiten einhergehen. Somit stellt das Anhalten dieser Expansion ein vielversprechendes therapeutisches Ziel dar. Jüngste Studien haben gezeigt, dass mithilfe von sogenannten Base Editoren punktgenaue Veränderungen in den CAG-Sequenzen eingefügt werden können, die die Wiederholungsstruktur unterbrechen, ohne die kodierte Aminosäuresequenz zu verändern.
Konkret bedeutet dies, dass beispielsweise bestimmte CAG-Codons in CAA umgewandelt werden, die ebenfalls Glutamin codieren, aber als natürliche Unterbrechungen im Wiederholungstrakt gelten und mit stabileren Sequenzen assoziiert sind. Diese durch base editing induzierten Unterbrechungen tragen nachweislich dazu bei, die somatische Instabilität in Patientenzellen entscheidend zu mindern. Langzeitkulturen von bearbeiteten Huntington-Fibroblasten zeigten wesentlich geringere Zunahmen der CAG-Repetitionslänge im Vergleich zu unbehandelten Kontrollzellen. Sogar eine leichte Verkürzung der Repeatlänge wurde beobachtet, was auf mögliche Kontraktionsprozesse hindeutet, die von den Editierungsmaßnahmen gefördert werden. In Tiermodellen der Huntington-Krankheit, wie den Htt.
Q111-Mäusen, konnte die In-vivo-Anwendung von Adeno-assoziierten Viren (AAV) eingesetzt werden, um die Base-Editoren direkt in das Gehirn zu schleusen. Über intrazerebroventrikuläre Injektionen in neugeborene Mäuse gelang eine effiziente Einführung von CAA-Unterbrechungen in den krankhaft erweiterten HTT-CAG-Trakten. In relevanten Hirnregionen wie dem Kortex und Striatum, die für die motorischen Symptome von HD eine Rolle spielen, reduzierte die Behandlung signifikant die somatischen Erweiterungen und stabilisierte die Repeat-Länge über Zeiträume von mehreren Monaten. Die wissenschaftlichen Erkenntnisse basieren auf hochpräzisen Analyseverfahren wie Next-Generation-Sequencing und speziellen Software-Tools zur Detektion von kleinen Basenveränderungen und variabler Repeat-Länge. Diese Methode ermöglicht es, die introduzierten Unterbrechungen sowie deren Auswirkungen auf die Genomstabilität quantitativ zu erfassen.
Neben dem Hauptvorteil der Minderung der somatischen Expansion wurde auch die Sicherheitsbilanz der Base-Editoren geprüft. Off-Target-Analysen zeigten, dass die Editierungen überwiegend spezifisch an den Zielsequenzen stattfanden. Weit verbreitete unerwünschte Veränderungen in proteinkodierenden Genen waren selten und betrafen meist synonyme Mutationen, die keine Auswirkungen auf die Proteinstruktur haben. Vereinzelt wurden potenziell relevante Veränderungen identifiziert, die jedoch in zukünftigen Studien noch genauer untersucht werden müssen, um Risiken abzuwägen. Die Erkenntnisse bestätigen, dass das gezielte Einführen von Unterbrechungen in CAG-Repeats eine vielversprechende molekulare Strategie gegen die Progression von Huntington darstellen kann.
Durch das Stabilisieren der Repeatlänge in somatischen Zellen könnte der Krankheitsverlauf verlangsamt oder sogar das Einsetzen klinischer Symptome hinausgezögert werden. Neben Huntington ist das Prinzip der sogenannten Repeat Interruptions auf weitere Erkrankungen anwendbar, die auf ähnlichen pathogenen Mechanismen beruhen, etwa Friedreich-Ataxie oder diverse spinocerebelläre Ataxien. Die universelle Anwendung von Base-Editing-Technologien, gekoppelt an effiziente und zielgerichtete Virustransportsysteme, eröffnet somit neue Perspektiven für die Gentherapie neurodegenerativer Erbkrankheiten. Natürlich bleiben Fragen zur Dauerhaftigkeit der Base-Editing-Effekte und der Systemtoxizität weiterer Forschung vorbehalten. Auch die Translation dieser vielversprechenden Ansätze in klinische Therapien erfordert umfangreiche präklinische Studien.
Die Optimierung der Vektorgrößen, die Minimierung möglicher Immunreaktionen auf virale Überträger sowie die Steigerung der Targeting-Präzision sind wichtige Entwicklungsziele. Neben AAV-Delivery wird aktuell an alternativen Zustellmethoden gearbeitet, die transienten oder zelltypspezifischen Expressionen der Editoren erlauben, um Off-Target-Effekte weiter zu reduzieren. Auch Fortschritte bei der Entwicklung von Base-Editoren mit verringerter unspezifischer Aktivität steigern die Sicherheit der Technologie. Auf zellulärer Ebene scheint die Mechanik der Repeat-Expansion ebenfalls komplex. Die Bindung und Aktivität von DNA-Reparaturmechanismen, die Bildung von nichtkanonischen DNA-Strukturen sowie transkriptionelle Belastungen sind Teil eines vielschichtigen Prozesses.
Die Interruptions können hierbei als Barriere wirken, die die Bildung solcher schädlicher Strukturen verhindern und damit Instabilitäten entgegenwirken. Damit wird die Bearbeitung der Trinukleotid-Wiederholungen nicht nur als Reparaturinstrument verstanden, sondern auch als Mittel, um die zugrunde liegenden molekularen Ursachen der somatischen Expansion auf natürliche Weise zu modulieren. Somit verbindet sich die Technik mit dem Prinzip, pathogene Mutationstypen durch stabile natürliche Varianten zu ersetzen. Zusammenfassend lässt sich festhalten, dass die Base-Editing-Technologie in Huntington-Patientenzellen und entsprechenden Tiermodellen eine wirksame Somatikstabilisierung der CAG-Repeats erzielen kann. Diese Fortschritte stellen einen bedeutenden Schritt Richtung personalisierter Genomtherapien für Huntington dar und können als Blaupause für weitere Expansionserkrankungen dienen.
Die Zukunft der Huntington-Therapie könnte somit maßgeblich durch solche präzisen genomischen Eingriffe geprägt sein, die nicht nur die Symptome lindern, sondern direkt an die genetische Wurzel der Erkrankung anknüpfen.