Die Erdnuss (Arachis hypogaea L.) stellt weltweit eine der bedeutendsten Ölpflanzen und Lebensmittelleguminosen dar. Ihre wirtschaftliche Relevanz spiegelt sich nicht nur in der Vielzahl ihrer Anwendungen wider, sondern auch in der Bedeutung von Merkmalen wie Samen- und Hülsengröße für Ertrag und Qualität. Trotz ihrer Wichtigkeit fehlte bislang ein ganzheitliches Verständnis der genetischen Grundlagen, die diese Merkmale beeinflussen. Eine brandaktuelle Studie liefert nun bahnbrechende Erkenntnisse durch eine umfassende Pangenomanalyse, die strukturelle Variationen im Erdnussgenom mit der Variation der Samengröße in Verbindung bringt und neue Perspektiven für die genetische Verbesserung eröffnet.
Die Komplexität des Erdnussgenoms entspringt seiner zweifachen Genomkopplung, da die kultivierte Erdnuss ein sogenannter Allotetraploid ist, entstanden durch die Hybridisierung zweier Wildarten, Arachis duranensis (AA) und Arachis ipaensis (BB). Diese Polyploidie bringt sowohl Herausforderungen als auch Chancen mit sich. Die enorme genomische Vielfalt innerhalb und zwischen Wild- und Kultivarsorten wirkt sich maßgeblich auf phänotypische Eigenschaften wie Samen- und Fruchtgröße aus. Bislang lag der Fokus größtenteils auf Einzelgenom-Referenzsequenzen, die oft Lücken und Unvollständigkeiten aufweisen und damit die Erforschung großskaliger struktureller Veränderungen erschweren. Die Pangenomforschung stellt eine innovative Herangehensweise dar, bei der verschiedene Genome einer Spezies zusammengeführt werden, um die gesamte genetische Vielfalt abzubilden.
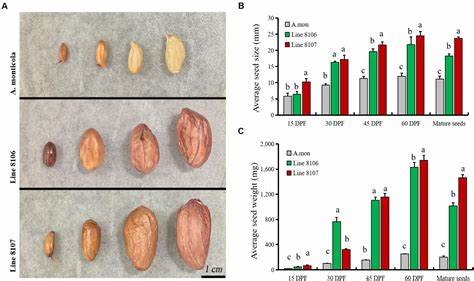
Im Fall der Erdnuss wurden acht hochwertige Genome – von Wild- und Kultivform – zusammengeführt und mit der Sequenzierung von 269 Zugängen mit großer phänotypischer Variation bei Samenmaßen kombiniert. So konnte ein umfassender Überblick gewonnen werden, der 50.097 Genfamilien unterschiedlicher Häufigkeit identifizierte, aufgeteilt in Kern-, verteilte und private Gene. Insbesondere die Analyse der strukturellen Variationen, darunter Einfügungen, Deletionen, Inversionen und Duplikationen mit Größen zwischen 50 und 100.000 Basenpaaren, zeigte eindeutige Unterschiede zwischen den Untergenomen A und B.
Bemerkenswert ist dabei, dass das A-Subgenom eine höhere Häufigkeit dieser Variationen aufweist, was auf unterschiedliche evolutionäre Selektionskräfte hindeutet. Die domestikationsbedingten Veränderungen verliefen asymmetrisch zwischen den Subgenomen. Während die Unterart A vor allem in den frühen Kultivierungsphasen starke Selektionsspuren zeigte, brachte Subgenom B später eine erhöhte Variabilität mit sich. Solche Unterschiede sind für erfolgreiche Züchtungsprogramme von großer Bedeutung, da sie Hinweise auf funktionsrelevante Gene und genetische Mechanismen liefern können. Eines der Highlights der Studie ist die Identifizierung eines wichtigen Gens, AhARF2-2, das eine Rolle als negativer Regulator der Samenentwicklung hat.
Eine spezifische 275-Basenpaar-Deletion führt zu einem Verlust der Interaktionsfähigkeit mit den Proteinen AhIAA13 und TOPLESS, welche normalerweise die Expression von Wachstum fördernden Faktoren wie AhGRF5 hemmen würden. Dadurch wird das Wachstum der Samen begünstigt, was insbesondere bei großen-ertragreichen Erdnussvarianten beobachtet wurde. Diese Entdeckung unterstreicht, wie einzelne strukturelle Variationen tiefgreifende Auswirkungen auf agronomisch relevante Merkmale haben können. Zusätzlich wurde ein weiteres Gen, AhCKX6, identifiziert, dessen Variation im 3'-UTR-Bereich mit einer verminderten Genexpression einhergeht und die Samengröße durch eine erhöhte Zytokininkonzentration in den sich entwickelnden Samen fördert. Cytokinine sind bekannte Pflanzenhormone, die das Zellwachstum und die Zellteilung regulieren und somit maßgeblich bei der Kontrolle von Organ- und Samenwachstum beteiligt sind.
Die Einsicht, dass solche hormonell bedingten Mechanismen durch strukturelle genomische Veränderungen beeinflusst werden, eröffnet neue Möglichkeiten, gezielt auf diese Signalwege einzuwirken. Die Forschung verdeutlicht auch den Einfluss von Transposons – besonders LTR-Retrotransposons und DNA-Transposons – auf die Dynamik des Erdnussgenoms. Ein erheblicher Anteil der strukturellen Variationen überschneidet sich mit repetitiven Elementen, die als Motoren der genetischen Variation fungieren. Solche Elemente können durch Einfügen oder Löschen von DNA-Sequenzen die Genregulation und damit auch phänotypische Traits beeinflussen. Ein weiterer zentraler Aspekt ist die Rolle der Genkopplung zwischen Ertrag und Krankheitsresistenz.
Während der Domestikation wurde beobachtet, dass Merkmale zugunsten höherer Erträge möglicherweise auf Kosten der Resistenz gegen diverse Pathogene gingen. Für Züchter bedeutet dies eine große Herausforderung, da die Balance zwischen diesen Eigenschaften erhalten oder sogar verbessert werden muss. Die Erkenntnis, dass bestimmte Genregionen, die sowohl Ertrags- als auch Resistenzgene beinhalten, als Tandem-Cluster in verwandten Accessions existieren, bietet Ansatzpunkte für beide Bereiche gleichzeitig. Die methodische Kombination modernster Sequenziertechnologien – Langzeitsequenzierung mittels Nanopore und PacBio HiFi, gekoppelt mit Hi-C für die Chromosomenstruktur – ermöglichte es, Lücken in der Genomassemblierung zu schließen und hochpräzise, nahezu vollständige Genomkarten zu erstellen. Diese Datenbasis ist für eine tiefgreifende Genomforschung und präzise Identifizierung von strukturellen Variationen unverzichtbar.
Insgesamt bietet die vorgestellte Pangenomanalyse eine umfassende genetische Ressource, die über bisherige Referenzgenome hinausgeht und das Verständnis der genetischen Kontrolle von Schlüsselfunktionen maßgeblich vertieft. Für die Züchtung von Erdnüssen eröffnet sie neue Wege, um sowohl traditionelle Merkmale wie Samen- und Fruchtgröße als auch Resistenz gegen Krankheiten effizient zu verbessern. Der Einsatz von genomischen Selektionstechnologien auf Basis dieser Erkenntnisse könnte zudem dazu beitragen, den Züchtungsprozess zu beschleunigen und den Bedarf an klassischen, zeitintensiven phänotypischen Selektionsverfahren zu reduzieren. Die Integration von genomischen Daten aus Pangenomen ermöglicht die präzisere Selektion von günstigen Allelen und öffnet Türen für molekulargenetische Optimierungen mithilfe von Geneditierung. Darüber hinaus schafft dieses Wissen wichtige Grundlagen für andere Leguminosen und polyploide Kulturpflanzen, die ähnliche genetische Strukturen und Domestikationsprozesse durchlaufen haben.
Die entwickelten bioinformatischen Werkzeuge und analytischen Ansätze lassen sich auf weitere Arten übertragen, um komplexe genetische Variationen besser zu verstehen und gezielt zu nutzen. Abschließend bleibt festzuhalten, dass die Studie einen bedeutenden Fortschritt im Bereich der Pflanzenforschung darstellt. Sie verbindet fortschrittliche Genomiktechnologien mit präziser phänotypischer Analyse und bietet damit eine Blaupause für zukünftige Forschungsbemühungen, die darauf abzielen, Ernährungssicherheit und nachhaltige Landwirtschaft weltweit zu fördern.