Das menschliche Genom, bestehend aus etwa zwei Metern DNA, ist hochkompakt in den winzigen Zellkernen von circa zehn Mikrometern Größe gefaltet. Diese Verpackung erfolgt nicht willkürlich, sondern nach einer strikt organisierten dreidimensionalen Architektur, die essenziell für die Regulierung der Genaktivität ist. Besonders in der Krebsforschung gewinnt die Erforschung dieser 3D-Genomstruktur zunehmend an Bedeutung, da sich zeigt, dass Veränderungen in der räumlichen Organisation des Genoms weitreichende Folgen für die Entstehung und das Fortschreiten von Tumoren haben können. Die neuesten Forschungen konzentrieren sich darauf, wie diese dreidimensionale Genomlandschaft in primären menschlichen Krebserkrankungen aussieht und welche Mechanismen der Genregulation daran beteiligt sind. Die Grundlage der 3D-Genomorganisation bilden chromosomale Kompartimente, sogenannte A- und B-Kompartimente, die große Segmente des Genoms umfassen und sich hinsichtlich chromatinischer Eigenschaften stark unterscheiden.
Die A-Kompartimente sind in der Regel aktiv, offen und genreich, während B-Kompartimente eher geschlossen und repressive chromatinische Merkmale aufweisen. Innerhalb dieser Kompartimente sind sogenannte Topologisch Assoziierte Domänen (TADs) organisiert, die als funktionelle Einheiten der chromosomalen Faltung angesehen werden. Diese Domänen ermöglichen Interaktionen innerhalb von Genabschnitten und schließen weitreichende Überschneidungen zwischen Bereichen meist aus. Auf einer noch feineren Ebene sind direkte Wechselwirkungen zwischen Enhancern und Promotoren von Genen entscheidend, um die zelltypspezifische Genexpression zu steuern. In humanen Krebserkrankungen kommt es häufig zu strukturellen Veränderungen des Genoms.
Diese sogenannten strukturellen Varianten umfassen Deletionen, Duplikationen, Inversionen und Translokationen, die nicht nur die Anzahl der Genkopien verändern, sondern auch die räumliche Organisation der Chromosomen dramatisch beeinflussen können. Solche Veränderungen können dazu führen, dass weit voneinander entfernte regulatorische Elemente neu angeordnet werden, sodass ein Onkogen durch einen bisher getrennten Enhancer aktiviert wird, ein Prozess, der als Enhancer-Rewiring bezeichnet wird. Dieses Phänomen spielt eine wichtige Rolle bei der Überexpression von Krebsgenen und beeinflusst maßgeblich die Entstehung der bösartigen Zellviefältigkeit. Ein besonders bemerkenswertes Beispiel dieser komplexen strukturellen Veränderung ist die extrachromosomale DNA (ecDNA). Diese zirkulären DNA-Moleküle sind eigenständige Einheiten außerhalb der eigentlichen Chromosomen und können große Kopienzahlen von Onkogenen tragen.
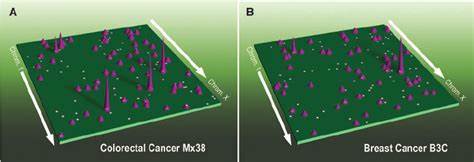
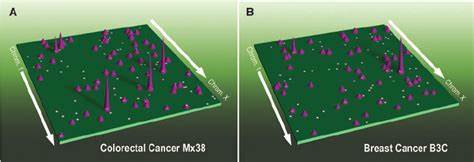
Sie bieten damit eine kraftvolle Möglichkeit zur Verstärkung der Krebsgenexpression und stehen in Zusammenhang mit aggressiven Tumorverläufen und schlechter Prognose. Die ecDNA hat den Vorteil gegenüber chromosomalen Kopien, dass sie räumlich flexibler interagieren kann, was zu einer extensiven Neukonfiguration von Enhancer-Promotor-Verbindungen und dadurch verstärkter Genaktivität führt. Um die dreidimensionale Genomlandschaft bei verschiedenen Krebsarten systematisch zu erfassen, wurden neuartige Methoden der Chromosomen-Konformationsforschung eingesetzt. Eine vielversprechende Technik ist die H3K27ac HiChIP, welche auf der Kombination von Chromatin-Immunpräzipitation mit chromosomaler Konformationsanalyse basiert. Sie ermöglicht eine proteingezielte Untersuchung der Dreidimensionalität des Genoms, indem sie zugleich Aktivität von Enhancern (über den H3K27ac-Histonmodifikationsmarker) und deren Interaktionen mit Zielgene erfasst.
Mit HiChIP konnten Forscher in aktuellen Studien 69 Tumorproben aus 15 verschiedenen primären menschlichen Krebsarten analysieren und damit ein detailliertes Bild der 3D-Genomarchitektur erzeugen. Die Ergebnisse der Analysen offenbarten drei Hauptmuster der Enhancer-Nutzung in Bezug auf über 100 Onkogene. Das erste Muster ist die statische Nutzung, bei der Enhancer unabhängig von der Krebsart konsistent mit einem Onkogen interagieren. Das zweite Muster beschreibt selektive Zugewinne bestimmter Enhancer in bestimmten Tumortypen. Das dritte und komplexeste beschreibt dynamisches Rewiring, bei dem die Verknüpfungen zwischen Enhancern und Promotoren je nach Tumorart oder sogar Patientenvarianten stark variieren.
Besonders wurde dies etwa beim MYC-Onkogen beobachtet, einem zentralen Regulator von Zellwachstum und Proliferation, dessen Enhancer-Umgebung unterschiedlich je nach Tumorart ausgeprägt ist. Darüber hinaus ermöglichte die Kombination der HiChIP-Daten mit tiefgehender Ganzgenomsequenzierung und Einzelzell-Chromatinzugänglichkeit eine verbesserte Zuordnung der Enhancerlandschaft in den verschiedenen Zelltypen des Tumormikromilieus. So konnten spezifische regulatorische Interaktionen in Tumorzellen sowie in Immun- und Stroma-Zellen isoliert werden. Besonders interessant sind dabei die enhancergen-Interaktionen im Zusammenhang mit Genen, die an der Immunflucht beteiligt sind, wie etwa CD274, das für PD-L1 codiert. PD-L1 ist ein wichtiger Immuncheckpoint, der von Tumoren genutzt wird, um die Immunabwehr zu unterdrücken.
Die Untersuchungen zeigen, dass nicht nur Tumorzellen, sondern auch myeloide Zellen im Mikromilieu spezifische Enhancer-Promotor-Verbindungen aufweisen, die die Expression von PD-L1 steuern. Wesentliche Erkenntnisse kamen auch aus der Analyse von nichtkodierenden somatischen Mutationen. Solche Mutationen, die in regulatorischen DNA-Abschnitten auftreten, können durch Schaffung neuer Transkriptionsfaktor-Bindestellen oder Verstärkung bestehender Enhancer deren Aktivität erhöhen und somit zur Aktivierung von Onkogenen führen. Mit Hilfe der Allelspezifität in HiChIP-Daten konnten Mutationen identifiziert werden, die in aktiven Enhancern mit höherer VAF (Variant Allele Frequency) vorliegen, was auf eine funktionelle Bedeutung dieser Mutationen hinweist. Beispiele wie eine somatische Punktmutation im Enhancer von FGFR1 in Blasenkrebs illustrieren den direkten Zusammenhang von Mutation, gesteigerter Enhancer-Aktivität, verstärktem Enhancer-Promotor-Kontakt und letztendlich erhöhter Onkogenexpression.
Die 3D-Genomforschung beleuchtet auch die Einflüsse komplexer struktureller Veränderungen auf die Genregulation bei Krebserkrankungen. Hierzu gehören chromothripsis, Brücke-Fusions-Brücke-Veränderungen und besonders ecDNA. Durch innovative bioinformatische Werkzeuge konnten die räumlichen neuen DNA-Verbindungen (so genannte Neoloops) und neu gebildete TADs (neoTADs) erkannt werden, die durch diese rearrangierten Strukturen entstehen. Interessanterweise zeigen verschiedene Klassen von amplifizierten DNA-Sequenzen unterschiedliche Muster ihrer Enhancer-Neuverknüpfung. Vor allem ecDNA ist dafür bekannt, ein besonders umfangreiches und dynamisches Netzwerk von Enhancer-Promotor-Wechselwirkungen zu fördern, das in mehreren Krebsarten beobachtet wurde.
Die Erkenntnisse über die 3D-Genomlandschaft bei Krebs stellen ein bedeutendes Forschungsfeld dar, das die klassische Sichtweise über Genregulation erweitert. Die Unterscheidung zwischen stabiler chromosomaler Organisation und flexiblen, zelltyp- oder tumorspezifischen Enhancer-Interaktionen ermöglicht ein tieferes Verständnis der molekularen Mechanismen von Tumorentstehung und Tumorheterogenität. Die Integration von räumlicher Genomfaltung mit Mutation, Genkopienzahlveränderung, Chromatinzugänglichkeit und Transkriptionsdaten schafft ein umfassendes Bild und kann bei der Identifikation individueller therapeutischer Ansatzpunkte helfen. Aus klinischer Sicht kann das Wissen um die Variabilität der Onkogenregulation mittels CNV und Enhancer-Aktivität dazu beitragen, genauer zu beurteilen, welche genetischen und epigenetischen Veränderungen entscheidend für die Tumorprogression sind. Beispielsweise lässt sich nun besser nachvollziehen, warum bei manchen Patienten die Überexpression von KRAS vor allem durch Gene-Kopienzahlveränderung erklärt wird, während bei anderen Onkogenen wie MET die Aktivität regulatorischer Elemente ausschlaggebender ist.
Diese differenzierte Betrachtung bietet die Chance für personalisierte Therapieansätze, die gezielt auf die zugrundeliegenden molekularen Störungen reagieren. Die Kombination aus HiChIP-Technologie und Single-Cell-Analyse ermöglicht auch, den Tumormikromilieu gezielt zu charakterisieren und so immunologische Zielstrukturen zu finden, die auf das Zusammenspiel zwischen Tumor- und Immunzellen abzielen. Dies ist besonders bedeutsam im Kontext der Immuntherapie, da hier die Funktion und Modulation von Immun-Checkpoints wie PD-L1 von zentraler Bedeutung ist. Abschließend lässt sich sagen, dass die Erforschung der dreidimensionalen Genomlandschaft in primären menschlichen Krebserkrankungen neue Horizonte im Verständnis der Krebsbiologie eröffnet. Die Anwendung moderner genomischer und epigenomischer Methoden macht es möglich, die komplexe Struktur und Funktion des Genoms in seiner räumlichen Dimension zu visualisieren und ihre Auswirkungen auf Krebsentwicklung und Erkrankungsverlauf zu analysieren.
Zukunftsträchtige Forschung wird sich darin erschöpfen, wie diese Erkenntnisse therapeutisch genutzt werden können, um gezielte Behandlungen zu entwickeln, die auf 3D-Genom-vermittelte Dysregulationen abzielen und somit bessere klinische Ergebnisse ermöglichen.