Die Erforschung der Ursprünge von Pandemieauslösern wie SARS-CoV und SARS-CoV-2 ist von entscheidender Bedeutung, um zukünftige Ausbrüche zu verhindern und besser zu kontrollieren. Beide Viren gehören zur Familie der Coronaviren und haben aufgrund ihrer Fähigkeit, vom Tier auf den Menschen überzuspringen, erhebliche globale Gesundheitskrisen verursacht. Die Übertragungswege und insbesondere die Vorfahren dieser Viren lassen sich in den weit verbreiteten Fledermauspopulationen nachvollziehen, die als natürliche Wirte vieler Coronaviren gelten. Dabei spielt nicht nur die geografische Herkunft eine wichtige Rolle, sondern auch die zeitliche Einordnung der viralen Varianten, die zur genetischen Evolution dieser gefährlichen Erreger geführt haben.Fledermäuse sind weltweit verbreitet und beherbergen eine Vielzahl von Viren.
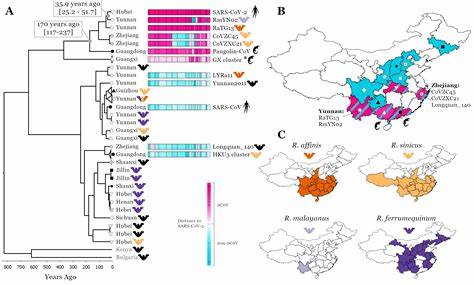
Besonders in Asien, vor allem in Süd- und Südostasien, aber auch in Teilen Afrikas und Europas, wurden zahlreiche Corona-ähnliche Viren in Fledermausarten nachgewiesen. Die genetische Verwandtschaft dieser Viren mit den Erregern von SARS und COVID-19 bietet wertvolle Hinweise auf die Ursprünge und auf potenzielle Übertragungsstationen zwischen Tier und Mensch. Forschungsergebnisse bestätigen, dass die direkten Vorfahren von SARS-CoV in Fledermäusen der Gattung Rhinolophus, auch bekannt als Hufeisennasenfledermäuse, gefunden wurden. Diese Fledermäuse sind aufgrund ihrer biologischen Eigenschaften und ihres Verbreitungsgebiets ein natürlicher Reservoir für eine Vielzahl von Coronaviren.SARS-CoV wurde erstmals im Jahr 2002 identifiziert und führte zu einer Epidemie, welche in China begann.
Wissenschaftler konnten Fledermausviren entdecken, die SARS-CoV genetisch sehr ähnlich waren, jedoch bereits Jahre zuvor in den Fledermäusen existierten. Dies zeigt, dass die Entstehung der viralen Vorläufer weit vor dem Ausbruch lag und sich über Jahrzehnte hinweg entwickelte. Die genetische Analyse weist darauf hin, dass Zwischenwirte, wie bestimmte Schleichkatzen (Civetkatzen), eine Schlüsselrolle bei der Übertragung auf den Menschen spielten. Dennoch bleibt der ursprüngliche Ursprung auf den Fledermäusen liegen, die das Virus über einen längeren Zeitraum beherbergten und genetisch diversifizierten.Der Fall von SARS-CoV-2 gestaltet sich ähnlich, aber mit zusätzlichen Komplexitäten.
Die Pandemie, die 2019 begann, wandelte binnen weniger Monate das Leben von Milliarden von Menschen weltweit. Frühere Studien konnten viele nahe Verwandte von SARS-CoV-2 in Fledermauspopulationen in der Provinz Yunnan, China, entdecken. Insbesondere das Virus RaTG13, ein Fledermausvirus, weist eine sehr hohe Übereinstimmung mit SARS-CoV-2 im Genom auf. Allerdings bestehen Unterschiede in kritischen Sequenzabschnitten, die für das Eindringen in menschliche Zellen verantwortlich sind. Diese Erkenntnis lässt vermuten, dass SARS-CoV-2 entweder durch einen Zwischenwirt, dessen Identität noch nicht abschließend geklärt ist, weiterentwickelt wurde, oder dass Fledermausviren unter natürlichen Bedingungen schon lange entsprechende Eigenschaften aufwiesen.
Neben China reichen die fledermausassoziierten Coronaviren geographisch über Länder wie Myanmar, Laos und Vietnam bis nach Südostasien hinein. Diese großräumige Verteilung unterstreicht die Notwendigkeit, die Fledermausvirome in einem regionalen Kontext zu betrachten, da verschiedene Subtypen und Varianten von Coronaviren in unterschiedlichen Populationen existieren. Die derzeitige Forschung zeigt, dass die genetische Durchmischung und Migration von Fledermäusen eine bedeutende Rolle für die Virenvielfalt und damit für das Risiko der zoonotischen Übertragung spielen. Dieses Zusammenspiel aus Biologie der Wirte, Umweltfaktoren und menschlichem Verhalten bestimmt maßgeblich das Entstehen und die Ausbreitung neuer Virusvarianten.Timing ist ebenso entscheidend für das Verständnis der Entwicklung dieser Viren.
Molekulare Uhren und phylogenetische Methoden erlauben es Forschern, die Mutationsraten und den Zeitpunkt, zu dem sich diese Viren voneinander getrennt haben, zu schätzen. Die Daten deuten darauf hin, dass die Systematik der SARS-ähnlichen Coronaviren in Fledermauspopulationen über Jahrzehnte stabil blieb, ehe einzelne Varianten die Fähigkeit erlangten, Menschen zu infizieren. Beim ursprünglichen SARS-CoV wird angenommen, dass der zoonotische Übergang Anfang der 2000er Jahre stattfand. Bei SARS-CoV-2 deuten molekulargenetische Berechnungen darauf hin, dass der Absprung auf den Menschen zwischen Mitte 2018 und Ende 2019 stattfand, was mit den ersten bekannten Fällen im Dezember 2019 übereinstimmt.Die Analyse der Ursprünge von SARS-CoV und SARS-CoV-2 zeigt eindrücklich, wie das Zusammenspiel von Ökologie, Evolution und menschlichem Einfluss zum Entstehen von Pandemien führen kann.