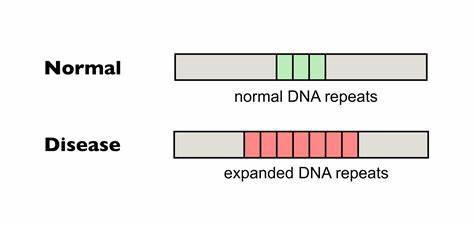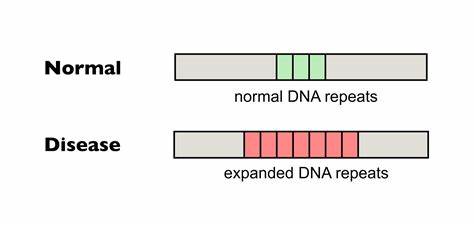
Die Huntington-Krankheit (HD) zählt zu einer Gruppe von schweren neurodegenerativen Erkrankungen, die durch krankhafte Ausdehnungen trinukleotidischer DNA-Wiederholungen verursacht werden. Im Zentrum dieser Pathologie stehen insbesondere verlängerte CAG-Abschnitte im Huntingtin-Gen (HTT), deren somatische Instabilität das Fortschreiten der Erkrankung maßgeblich beeinflusst. Jüngste Forschungen konzentrieren sich zunehmend auf die gezielte Veränderung dieser repetitiven DNA-Sequenzen, um die Expansion der CAG-Wiederholungen in betroffenen Zellen zu bremsen oder zu verhindern. Diese Entwicklungen eröffnen neue therapeutische Perspektiven und könnten die Lebensqualität von Patienten nachhaltig verbessern. Trinukleotid-Wiederholungserkrankungen, zu denen neben Huntington auch Friedreich-Ataxie und diverse Spinocerebelläre Ataxien zählen, resultieren aus der instabilen Ausdehnung kurzer DNA-Repeatsequenzen.
Diese wachsenden Wiederholungen führen zu Fehlfunktionen der betroffenen Gene und letztlich zu neurodegenerativen Schäden. Für HD stellt die Länge der CAG-Wiederholungen die wichtigste prädiktive Variable für Krankheitsbeginn und -schwere dar. Dabei ist nicht nur die erbliche Länge entscheidend, sondern auch die dynamische Expansion der Wiederholungen in somatischen Geweben während des Lebens des Patienten. Basierend auf dieser Erkenntnis zielen neue molekulare Therapieansätze darauf ab, die sogenannte somatische Instabilität zu reduzieren. Einer der innovativsten Ansätze ist die Verwendung von Baseneditierung, einer CRISPR/Cas9-basierten Technologie, die präzise einzelne Nukleotide in der DNA verändern kann, ohne Doppelstrangbrüche zu induzieren.
Im Gegensatz zur klassischen CRISPR-Technologie eröffnet die Baseneditierung die Möglichkeit, einzelne Basen gezielt umzuwandeln und somit genetische Sequenzen durch subtile Veränderungen zu stabilisieren. Im Kontext der Huntington-Krankheit wurde gezeigt, dass die Einfügung von sogenannten Interruptions—das sind einzelne Basenveränderungen, die die Wiederholungskette unterbrechen—die Instabilität der CAG-Trakte reduzieren kann. Genauer gesagt wurden CAG-Sequenzen mittels Cytosin-Baseneditoren in CAA-Tripletts umgewandelt, die zwar ebenfalls für Glutamin kodieren, aber strukturell die Bildung von schädlichen DNA-Haarpinnekonformationen, welche Expansionen fördern, unterbrechen. Studierende und erfahrene Wissenschaftler fanden heraus, dass die Anwesenheit solcher Interruptions in menschlichen und tierischen Modellen mit einer signifikanten Verringerung der somatischen Repeat-Expansion verbunden ist. Die praktische Umsetzung erfolgte unter anderem durch den Einsatz von Adeno-assoziierten Viren (AAVs) vom Serotyp 9, welche die Baseneditor-Gene gezielt in neuronale Gewebe, insbesondere in das Gehirn von Huntington-Mausmodellen, einbrachten.
Durch neonatale intrazerebroventrikuläre Injektionen konnte eine effiziente Verbreitung und Expression der Editoren sichergestellt werden, was zur Einführung vieler Interruptions innerhalb der pathogenen CAG-Wiederholung führte. Als Ergebnis zeigte sich nicht nur eine Verringerung der somatischen Expansion der CAG-Wiederholungen in Hirngeweben, sondern auch eine teilweise Stabilisierung oder leichte Kontraktion der Repeat-Länge, ein möglicher Schlüssel zur Verzögerung oder Abschwächung des Krankheitsverlaufs. Neben der Verbesserung der molekularen Marker für HD legen diese Befunde nahe, dass die somatische Instabilität der CAG-Repeats ein therapeutisch relevanter Zielpunkt ist. Die Einführung von Repeat-Unterbrechungen durch Baseneditierung könnte die Entwicklung neuer Behandlungsmöglichkeiten prägen, die nachhaltig auf die Genom-Stabilität abzielen, statt allein symptomatisch zu wirken. Die hohe Sequenzähnlichkeit der CAG-Abschnitte im HTT-Gen mit anderen genomischen Regionen führte bei der Entwicklung der Baseneditierer zu der Herausforderung, off-target Effekte zu minimieren.
Umfangreiche Analysen mittels CIRCLE-seq (eine Methode zur in vitro-Off-target-Detektion) und hochauflösender Ganzgenomsequenzierungen enthüllten, dass die meisten Off-target-Veränderungen entweder in nicht-kodierenden oder synonymen Regionen auftreten, die keine schädlichen Auswirkungen auf die Proteinfunktion erwarten lassen. Dennoch ist eine weitere Untersuchung und langfristige Überwachung dieser Effekte für eine künftige klinische Anwendung unerlässlich. Zusätzlich zu Huntington wurden vergleichbare Strategien bei Friedreich-Ataxie untersucht, bei der eine Expansion von GAA-Tripletts im FXN-Gen ursächlich ist. Auch hier zeigte die Anwendung von Adenin-Baseneditoren, die Synonyme oder Interruptions in den GAA-Trakten erzeugen, eine Reduzierung der somatischen Repeat-Expansion mit einhergehendem Anstieg der FXN-Genexpression. Diese parallelen Erkenntnisse verdeutlichen den universellen Wert der Baseneditierung bei der Behandlung verschiedener TNR-Erkrankungen.
Die Entwicklung präziser und effektiver Baseneditierungswerkzeuge, die auf die speziellen Bedürfnisse von TNR-Erkrankungen zugeschnitten sind, stellt einen bedeutenden Fortschritt in der Genommedizin dar. Die bestehende Forschung kombiniert molekulargenetische Methoden mit fortgeschrittenen viralen Vektoren und Zellkultur- sowie Tiermodellstudien, was ein umfassendes Verständnis der Pathogenese und möglicher Interventionen ermöglicht. Zukünftige Herausforderungen liegen unter anderem in der Optimierung der Editoren hinsichtlich Effizienz und Spezifität, der Erweiterung der Zielgewebe jenseits des zentralen Nervensystems und der Evaluierung möglicher Immunreaktionen auf virale Vektoren oder die Editoren selbst. Ferner ist die Entwicklung von Therapieschemata, die wiederholte oder späte Behandlungen bei symptomatischen Patienten ermöglichen, von hoher Relevanz. Die Integration der eingeführten Interruptions in humane Patienten-Genome stellt zudem ein neues Paradigma in der Gentherapie dar.
Statt der vollständigen Korrektur der pathogenen Mutation ermöglichen solche subtile Sequenzvariationen eine stabilisierende Wirkung auf die ominöse somatische Instabilität, die viele der neurodegenerativen Wirkungen mitverursacht. Insgesamt demonstrieren die Fortschritte in der gezielten Baseneditierung, dass die aufwendige Bearbeitung repetitiver DNA-Sequenzen nicht nur technisch machbar ist, sondern auch eine vielversprechende Strategie darstellt, um wesentliche krankheitsfördernde Mechanismen bei Huntington und verwandten Erkrankungen zu verlangsamen oder zu unterbrechen. Dies ebnet den Weg für zukünftige klinische Studien und potenziell wirksame Behandlungsformen, die auf molekularer Ebene das Fortschreiten dieser bislang unheilbaren Erkrankungen bremsen können.