Die Erforschung der menschlichen Abstammungsgeschichte hat im letzten Jahrzehnt enorme Fortschritte gemacht, insbesondere durch moderne genetische Analysemethoden. Insbesondere die Anwendung eines strukturierten Koaleszenzmodells – einer komplexen statistischen Methode zur Rekonstruktion von Abstammungslinien anhand genetischer Daten – hat die Tiefe und Komplexität unserer gemeinsamen Herkunft noch deutlicher gemacht. Dieses Modell geht weit über die traditionelle Annahme einer einzigen, ungeteilten Urpopulation hinaus und zeigt, dass moderne Menschen von mindestens zwei alten, genetisch voneinander getrennten Populationen abstammen, die vor etwa 1,5 Millionen Jahren divergierten und sich vor etwa 300.000 Jahren mischten. Diese neue Erkenntnis transformiert unser Verständnis von menschlicher Evolution, genetischer Vielfalt und der Entstehung unserer Spezies.
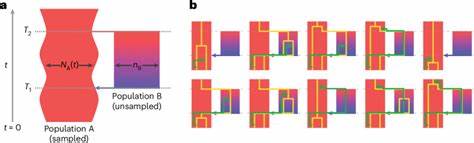
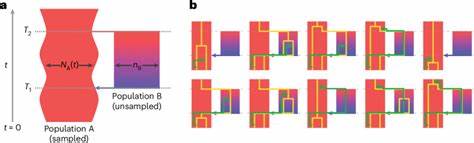
Das strukturierte Koaleszenzmodell „cobraa“ Die Methode, die diese Entdeckungen ermöglicht hat, nennt sich cobraa (coalescence-based reconstruction of ancestral admixture). Sie baut auf dem Prinzip der Koaleszenz auf, einem Ansatz, der Daten von einzelnen Genomsequenzen nutzt, um zu rekonstruieren, wie sich Populationen im Laufe der Zeit geteilt und wieder zusammengeführt haben. Anders als traditionelle Modelle, die eine kontinuierliche Mischung innerhalb einer einheitlichen Population annehmen, integriert cobraa modellhaft eine „Puls“ -Struktur – das heißt, eine einmalige oder wenige Isolations- und Vermischungsevents zwischen zwei Populationen A und B. In diesem Modell spaltet sich eine ursprüngliche Population in zwei getrennte Linien auf, die über einen langen Zeitraum isoliert bleiben, ehe sie innerhalb eines bestimmten Zeitfensters wieder verschmelzen. Diese Modellierung erlaubt es, nicht nur die Koaleszenzraten – also den Zeitpunkt der letzten gemeinsamen Vorfahren – zu betrachten, sondern auch die Übergangswahrscheinlichkeiten zwischen verschiedenen Koaleszenzzeiten entlang des Genoms, wodurch sich feinere Strukturen und vergangene Nutzungs- und Vermischungsprozesse erkennen lassen.
Dadurch lassen sich deutlich differenziertere Rückschlüsse auf alte Populationen, deren Größe, deren Trennung und erneute Verbindung ziehen. Die menschliche Abstammungsgeschichte neu denken Die Anwendung von cobraa auf genetische Daten aus dem Großprojekt 1000 Genomes sowie dem Human Genome Diversity Project zeigte konsistent, dass alle modernen Menschen genetisches Material von zwei großflächig divergenten Populationen geerbt haben. Diese Populationen trennten sich vor rund 1,5 Millionen Jahren und vermischten sich später vor circa 300.000 Jahren wieder, einer Periode, die mit dem Auftreten früher anatomisch moderner Menschen zusammenfällt. Rund 80 Prozent der heutigen menschlichen DNA lassen sich auf die eine Hauptpopulation (A) zurückführen, während etwa 20 Prozent von der zweiten, kleineren Population (B) stammen.
Interessanterweise wurde unmittelbar nach der Trennung der beiden Populationen ein starker demographischer Engpass in der vorherrschenden Population festgestellt. Ein solcher Flaschenhals – also eine drastische Reduktion der Populationsgröße – hat bedeutende evolutionäre Folgen, denn er kann genetische Vielfalt verringern und bestimmte Merkmale fixieren oder entfernen. Danach begann die Populationsgröße wieder zu wachsen, bis es zur besagten Wiedervereinigung der Populationen kam. Die Tatsache, dass das Genmaterial der Minderheit (Population B) heute in vielen Genomregionen als mit nachteiligen Effekten verbunden sichtbar ist, legt nahe, dass nach der Vermischung eine natürliche Selektion gegen bestimmte Allele dieser Population wirkte. Zudem wurde festgestellt, dass Segmente des Genoms, die der Mehrheitsbevölkerung zugeordnet werden, eine stärkere Nähe zu den Archaischen Arten Neanderthaler und Denisovaner aufweisen, was darauf hinweist, dass Population A auch deren gemeinsame Vorfahren mit modernen Menschen umfasst.
Die Auswirkungen auf unser Verständnis von menschlicher Evolution Diese Ergebnisse zwingen Wissenschaftler dazu, die einfache Vorstellung eines einzigen, einheitlichen Ursprungspopulationmodells für Homo sapiens zu überdenken. Stattdessen zeichnet sich ein Bild ab, in dem die Abstammung moderner Menschen aus komplexen Prozessen von tief geteilten und wieder zusammengeführten Populationen besteht. Die angewandte Methode zeigt, dass die genetische Geschichte des Menschen eine lange Phase isolierter Entwicklung beinhaltete, bevor eine bedeutende Vermischung stattfand, die die Grundlage der heutigen genetischen Vielfalt bildete. Diese Mischung aus getrennten Linien vor etwa 300.000 Jahren fällt zeitlich mit wichtigen paläoanthropologischen Funden: So erscheinen erste anatomisch moderne menschliche Fossilien etwa zu dieser Zeit, unter anderem in Jebel Irhoud (Marokko).
Das genetische Modell ergänzt die archäologischen Befunde und zeigt eine Kombination aus getrennter Evolution und Hybridisierung, welche die menschliche Vielfalt prägte. Insbesondere erklärt das Vorhandensein arkaischen Erbguts nicht nur die genetische Variation außerhalb Afrikas, die durch Neanderthaler- und Denisovanerdeleationen bekannt ist, sondern auch tiefere, bis dato weniger verstandene Afrikanische demographische Ereignisse. So ist die genetische Geschichte afrikanischer Populationen in dieser Hinsicht vielschichtiger, was neue Ansätze und Datenerhebungen erfordert. Genetische Signale in funktionellen Genomregionen Darüber hinaus bietet das Modell Einblicke in die evolutionären Kräfte, die nach der Durchmischung wirkten. So korrelieren die Abschnitte, die von der Minderheitspopulation stammen, mit geringerem Abstand zu Protein-codierenden Regionen, was auf negative Selektion hinweist.
Gleichzeitig zeigen Bereiche, die von der Mehrheitspopulation stammen, besondere Divergenzen zu archaischen Spezies, was wiederum die Verwandtschaftslinien innerhalb Homo-Neanderthaler-Denisovan-Komplex beleuchtet. Genomweite Skalenanalysen enthüllen zudem eine spezifische Anreicherung von introgressiven Genen in Bereichen, die neuronale Funktionen betreffen, etwa bei Zelladhäsionsprozessen oder Neurotransmission. Dies deutet darauf hin, dass genetische Variationen aus diesen alten Populationen möglicherweise Einfluss auf die Entwicklung und Funktion des Nervensystems hatten, wodurch evolutionäre Vorteile vermittelt wurden. Methodische Vorteile und Grenzen Die Stärke von cobraa liegt in der Kombination von Sequenzdaten mit einem präzisen Überlebensmodell für Koinzidenzzeitpunkte entlang des Genoms. Dadurch lassen sich demographische Ereignisse identifizieren, die mit anderen gängigen Methoden verborgen blieben oder falsch interpretiert wurden.
So konnte gezeigt werden, dass rein auf Populationengrößenveränderungen basierende Modelle – etwa PSMC – gewisse Populationsteilungen nicht korrekt abbilden oder als vermeintliche Größenänderungen fehlinterpretieren. Trotzdem bestehen Beschränkungen: So unterstellt das Modell eine isolierte Pulsstruktur und konstante Populationsgröße für die nicht-probierte Population B zwischen Trennung und Vermischung. Ebenso setzt cobraa auf Annahmen zur Neutralität und stetigen Mutations- und Rekombinationsraten. Komplexere historische Szenarien mit wiederkehrendem Genfluss oder kontinuierlichen Größenänderungen sind damit nicht vollständig erfassbar. Zudem ist die Genauigkeit bei kleinen Anteilen an introgressivem Erbgutoberhalb der etwaigen 5-Prozent-Marke am höchsten.
Für künftige Forschungen ergeben sich daraus Perspektiven, etwa durch Integration weiterer genetischer Konfliktlinien oder durch Kombination mit Methoden, die eine größere Zahl von Genome aus mehreren Individuen analysieren, um etwa wiederkehrenden Genfluss besser nachzuzeichnen. Erweiterung auf andere Spezies Das cobraa-Modell wurde auch erfolgreich auf andere Tierarten angewandt, einschließlich Delfinen, Gorillas und Schimpansen. Dabei zeigten sich je nach Art unterschiedliche Muster von abgestuften strukturellen Ereignissen in der Stammesgeschichte, die teilweise mit bisherigen Forschungsergebnissen kompatibel sind, teils aber neue Hypothesen generieren. Bei anderen Spezies, wie der parti-coloured Fledermaus, wurde kaum Anhalt für tiefe Populationsteilungen gefunden. Dies unterstreicht die breite Anwendbarkeit der Methode und ihren potenziellen Beitrag zur vergleichenden Evolutionsbiologie, mit Rückschlüssen darauf, wie diverse Tierarten mechanistisch auf Umweltveränderungen und Bevölkerungsdruck reagierten.
Wissenschaftliche und gesellschaftliche Relevanz Das Verständnis, dass unsere Abstammung aus einem komplexen Geflecht alter Populationen stammt, hat weitreichende Implikationen. Es verändert nicht nur akademische Sichtweisen, sondern prägt auch unser kulturelles Selbstbild. Die genetische Geschichte zeigt, dass genetische Vielfalt und Verhandlungen um Herkunft tief im Kern unserer Spezies liegen. Die Erkenntnisse unterstützen die Idee eines afrikanischen Ursprungskontinents für alle Menschen, jedoch nicht als statische Einheit, sondern als dynamische Landschaft mit vielfältigen Wechselwirkungen. Solche Nuancen sind grundlegend für ein modernes Verständnis von Evolution, Gesundheit, Erbkrankheiten und der zukünftigen Nutzung von genomischen Ressourcen.
Zusammenfassung Das strukturierte Koaleszenzmodell cobraa offenbart eine weit zurückreichende geteilte genetische Geschichte aller modernen Menschen. Stimmen über zwei alte, getrennt evoluierte Populationen wurden identifiziert, die sich vor ca. 1,5 Millionen Jahren voneinander trennten und vor rund 300.000 Jahren wieder Vermischung fanden. Diese Entdeckung kontrastiert mit älteren Modellen einer stetig vermischten Population und vertieft das Bild unserer komplexen Herkunft.
Die Kombination von hochauflösenden Genomdaten mit fortschrittlicher Modellierung markiert einen Meilenstein für die Paläogenetik und bietet die Grundlage, um weitere Fragen zur menschlichen Evolution und der Rolle archaischer Gene zu klären. Zugleich eröffnet die Methode Potenzial zur Anwendung in der Erforschung evolutionärer Dynamiken bei anderen Wirbeltierarten. In Zeiten, in denen unsere biologische Geschichte neue Perspektiven offenbart, wird das Bewusstsein für unsere gemeinsame Herkunft gestärkt und die wissenschaftliche Basis für weitere Entdeckungen gelegt, die unser Verständnis der biologischen Vielfalt und des menschlichen Wesens weltweit bereichern werden.