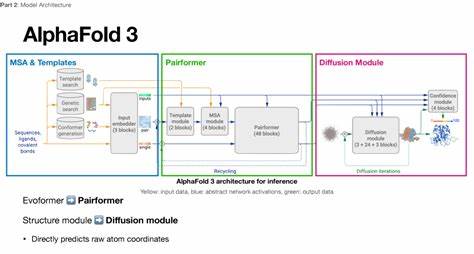
Die Vorhersage von Proteinstrukturen spielt eine zentrale Rolle in der modernen Biowissenschaft, da sie das Verständnis von biologischen Prozessen auf molekularer Ebene erheblich vertieft. Mit der Veröffentlichung von AlphaFold 3 wurde ein weiterer Meilenstein gesetzt, der präzisere und schnellere Vorhersagen ermöglicht. Besonders spannend ist dabei die Entwicklung einer hackbaren Version von AlphaFold 3, die ohne die früher üblichen Docker-Container und umfangreiche Mehrsequenz-Alignments (MSAs) auskommt. Dieses neue Konzept erleichtert nicht nur den Zugang zur Technologie, sondern senkt auch erheblich die technischen und infrastrukturellen Hürden für die Nutzung der Software. AlphaFold 3 ist die neueste Iteration des von DeepMind entwickelten AlphaFold-Systems zur präzisen Vorhersage von Proteinstrukturen.
Die ursprünglichen Versionen waren geprägt von hohem Rechen- und Speicherbedarf, insbesondere durch die Notwendigkeit, große Datenbanken mit MSAs herunterzuladen und zu verarbeiten. Dabei handelte es sich oft um mehrere hundert Gigabyte, die erst lokal bereitgestellt werden mussten. Außerdem erfolgte der Betrieb meist in Docker-Containern, um die komplexen Abhängigkeiten und Bibliotheken zu verwalten. Diese Kombination konnte für viele Nutzer, insbesondere mit begrenzter Hardware oder ohne umfassende IT-Unterstützung, eine große Hürde darstellen. Das hackbare AlphaFold 3 adressiert diese Einschränkungen direkt.
Durch den Verzicht auf Docker als Containerisierungsmethode und die Möglichkeit, auf MSAs komplett zu verzichten oder diese manuell und minimalistisch zu gestalten, wird der Einstieg in die Proteinstrukturvorhersage deutlich barriereärmer. Stattdessen kann das Programm nun leichter auf einzelnen Laptops oder auch kleineren GPU-Servern installiert und ausgeführt werden. Die Installation läuft reibungslos über Conda-Umgebungen ab, und die notwendigen Abhängigkeiten fokussieren sich auf JAX, Haiku und andere essentielle Python-Bibliotheken für maschinelles Lernen und numerische Berechnungen. Ein weiterer großer Vorteil ist die Reduzierung des Speicherbedarfs und der Datenvorhaltung. Während bisher massive Datenquellen notwendig waren, genügt nun eine kompakte Modell-Checkpoint-Datei, die unter den entsprechenden Nutzungsbedingungen von DeepMind bezogen werden kann.
Damit entfällt der Download und die lokale Speicherung der umfassenden MSA-Datenbanken. Forschungseinrichtungen oder einzelne Wissenschaftlerinnen und Wissenschaftler profitieren somit von einer deutlich schnelleren Setup-Zeit und benötigen keine umfangreiche IT-Infrastruktur mehr. Die leichte Modularität und offene Struktur des Codes erlauben darüber hinaus effektives Experimentieren und individuelle Anpassungen, was für methodische Entwicklungen und innovative Anwendungen von großem Wert ist. Der Verzicht auf MSAs bedeutet jedoch nicht zwingend eine Verschlechterung der Ergebnisqualität. AlphaFold 3 wurde dahingehend optimiert, auch bei minimalen bis keinen MSAs zuverlässige Vorhersagen zu liefern.
Dies ist besonders relevant für neuartige oder seltene Proteine, für die keine umfangreichen homologen Sequenzdaten existieren. Somit erschließt die Software neue Anwendungsfelder, von der Grundlagenforschung bis hin zur Wirkstoffentwicklung, wo schnelle und flexible Strukturvorhersagen essenziell sind. Die Nutzenden können die Vorhersagen direkt über Python-Skripte oder Jupyter-Notebooks steuern, was die Integration in bestehende Workflows und die Visualisierung der Ergebnisse erleichtert. Zudem ermöglichen diverse Konfigurationsoptionen, dass nur der wesentliche Teil des Verarbeitungsprozesses ausgeführt wird. Beispielsweise kann die Datenpipeline, welche normalerweise genetische und Templatesuchen durchführt, deaktiviert werden, wenn dies nicht benötigt wird.
Dies führt zu einer beträchtlichen Zeitersparnis bei der Ausführung, wodurch noch schneller 3D-Modelle generiert werden können. Die Performance des hackbaren AlphaFold 3 ist beeindruckend. Durch die Nutzung moderner GPU-Technologien und Frameworks wie JAX mit optimierten Triton-Kernels profitiert die Software von hoher Rechengeschwindigkeit und guter Ressourcennutzung. Auch für Nutzer mit älteren GPUs gibt es entsprechende Anpassungen in den Umgebungsvariablen, um eine möglichst störungsfreie und effiziente Ausführung sicherzustellen. Trotz der gewissen Komplexität im Hintergrund sind die Installationsanleitungen verständlich und gut dokumentiert, so dass selbst Einsteiger ohne tiefgreifendes Know-how in der Softwareentwicklung die Möglichkeit haben, AlphaFold 3 produktiv einzusetzen.
Ein weiterer bemerkenswerter Aspekt ist die rechtliche Komponente. Die Modellparameter von AlphaFold 3 sind ausschließlich direkt von Google DeepMind erhältlich und nur unter strengen Nutzungsbedingungen einsetzbar. Dies stellt sicher, dass die Verbreitung und Anwendung der Software in einem ethisch und rechtlich kontrollierten Rahmen erfolgt. Die Entwickler betonen, dass die Vorhersagen mit Vorsicht zu interpretieren sind und keine klinische Entscheidungsgrundlage darstellen. Dies soll Verantwortungsbewusstsein bei der Nutzung fördern und Risiken minimieren.
Durch die Entwicklung des hackbaren AlphaFold 3 ohne Docker und MSA-Zwang haben Forscher und Entwickler nun Zugang zu einer flexiblen, ressourcenschonenden und leicht verständlichen Plattform. Diese ermöglicht es nicht nur, Proteinstrukturen effizienter vorherzusagen, sondern auch die Algorithmen und Methoden nach Belieben zu modifizieren oder besser zu verstehen. Für wissenschaftliche Projekte, Bildungszwecke oder industrielle Anwendungen bietet dieses Modell eine hervorragende Grundlage, um Erkenntnisse im Bereich der Strukturbiologie schnell und unabhängig von großer Infrastruktur zu gewinnen. Die Kombination aus moderner Deep-Learning-Technologie, optimierter Softwarearchitektur und der bewussten Reduktion auf das Wesentliche stellt einen großen Fortschritt dar. Besonders im Zuge der zunehmenden Digitalisierung von Forschungsprozessen und dem Bedürfnis nach schneller Verfügbarkeit von Daten und Ergebnissen setzt das hackbare AlphaFold 3 einen neuen Standard.