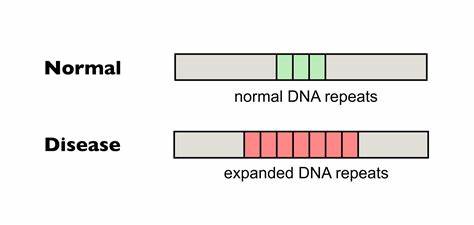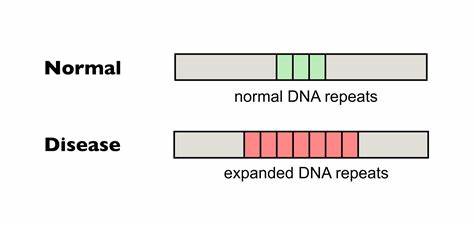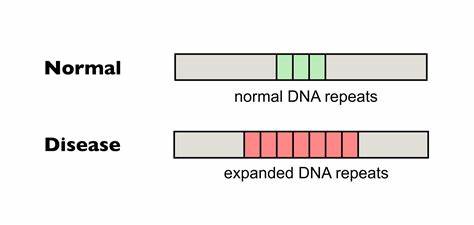
Die Huntington-Krankheit (HD) ist eine verheerende neurodegenerative Erkrankung, die durch die Expansion von CAG-Trinukleotidwiederholungen im Huntingtin-Gen ausgelöst wird. Diese veränderten Wiederholungen führen zu einer pathogenen Verlängerung der Polyglutamin-Kette, die giftige Proteinaggregate verursacht und die neuronale Funktion erheblich beeinträchtigt. Ein zentrales Merkmal der Erkrankung ist die somatische Instabilität der CAG-Tripletts, also deren wiederholte Expansion in verschiedenen Körperzellen, insbesondere im zentralen Nervensystem. Die somatische Expansion erhöht den Schweregrad und beschleunigt den Krankheitsausbruch, wodurch ein Therapieansatz entsteht, der darauf abzielt, diese Expansionen zu hemmen oder zu korrigieren. Neueste Forschungen konzentrieren sich darauf, die Repetitionen direkt im Genom zu verändern, um die pathogene Kettenlänge zu stabilisieren oder zu verkürzen.
Eine vielversprechende Technologie ist das sogenannte Base Editing, mit dem einzelne Nukleotide gezielt ohne Bruch der DNA-Doppelhelix umgewandelt werden können. Diese Methode ermöglicht es, die rein aus CAG-Wiederholungen bestehenden Sequenzen durch gezielte Veränderungen zu unterbrechen, indem beispielsweise einige CAG-Sequenzen in CAA umgewandelt werden. Obwohl beide Codons für Glutamin kodieren, bieten Unterbrechungen wie CAA eine signifikante Stabilitätssteigerung der Repeat-Motive, wodurch die Tendenz zur somatischen Expansion verringert wird. In Zellkulturmodellen von Huntington konnten Wissenschaftler zeigen, dass die Anwendung von Cytidin-Base-Editoren erfolgreich CAG-Trinukleotide in CAA umwandelt. Dies führte zu einer deutlichen Reduktion der somatischen Expansion über mehrere Zellteilungen hinweg.
Die Einführung dieser Unterbrechungen stellt somit eine vielversprechende Strategie dar, die dem Krankheitsverlauf entgegenwirken kann, indem die instabilen Repeats auf einen stabileren Status zurückgesetzt werden. Darüber hinaus wurde die Technik erfolgreich in vivo in Mausmodellen mit humanisiertem Huntingtin-Gen getestet. Über die virale Zustellung von Adeno-assoziierten Viren (AAV) gelang es, gezielt Nervenzellen im Gehirn zu transduzieren und die Pathogenese durch die Reduktion von Repeat-Erweiterungen zu verlangsamen. Die Ergebnisse zeigten nicht nur eine Verringerung der somatischen Erweiterungen in behandelten Hirnregionen wie Kortex und Striatum, sondern auch eine Stabilisierung und teilweise Kontraktion der bereits pathologisch verlängerten CAG-Tracts. Neben der Effektivität sind die Sicherheit und die möglichen Off-Target-Effekte von Base Editing zentral für eine zukünftige Therapiedurchführung beim Menschen.
Umfangreiche genomweite Analysen belegten, dass die meisten durch die Base Editoren induzierten Veränderungen im Genom entweder im nichtkodierenden Bereich liegen oder zu stillen Mutationen in proteinkodierenden Regionen führen. Solche stillen Mutationen verändern die Aminosäuresequenz des Huntingtin-Proteins nicht und sind wahrscheinlich ohne schädliche Konsequenzen. Dennoch ist die Erforschung möglicher Nebenwirkungen unerlässlich, um das Risiko bestmöglich zu minimieren. Der therapeutische Nutzen der Reduktion somatischer Repeat-Expansionen liegt vor allem darin, dass der Zeitpunkt des Krankheitsausbruchs maßgeblich durch die Länge der CAG-Wiederholung bestimmt wird. Je länger die reine Wiederholungssequenz, desto früher setzt die neurodegenerative Symptomatik ein.
Interruptions wie die natürliche Variation CAA innerhalb der CAG-Tracts korrelieren daher mit einem verzögerten Ausbruch und milderen Verläufen. Die gezielte Erzeugung solcher Unterbrechungen durch Base Editing könnte die Progression also spürbar hinauszögern. Die Bedeutung dieser Forschung ist vor allem im Kontext fehlender kausaler Therapien für Huntington zu sehen. Bislang konzentrieren sich Behandlungsoptionen hauptsächlich auf symptomatische Linderung. Die Möglichkeit, direkt in das genetische Grundproblem der Krankheit einzugreifen und die pathologische Repeat-Expansion zu stoppen oder umzukehren, eröffnet revolutionäre Perspektiven.
Neben Huntington erlaubt die Base Editing-Methode auch Ansätze für weitere trinukleotidbasierte Erkrankungen wie Friedreich-Ataxie, bei denen ähnliche Mechanismen der Repeat-Expansion eine Rolle spielen. Hier wurden unter anderem die GAA-Repeat-Regionen in patientenspezifischen Zellen bearbeitet, was zu einer Stabilisierung der Wiederholung und einer verbesserten Genexpression führte. Die Herausforderungen für die Übertragung der Base Editing-Technologie in klinische Anwendungen liegen in der effizienten und sicheren Zustellung des Editors in relevante Zelltypen, insbesondere Neuronen, sowie der Verhinderung langfristiger unerwünschter Effekte. Die aktuelle Forschung nutzt Optimierungen der viralen Vektoren und transient wirksame Editoren, um diese Hürden zu überwinden. Zusammenfassend stellt die genetische Modifikation der CAG-Repeats mittels Base Editing einen vielversprechenden Ansatz dar, um die somatische Instabilität bei Huntington zu verringern und somit die Krankheit progression aufzuhalten oder zu verzögern.
Die bisherigen Ergebnisse in Patientenzellen und Modellorganismen sind vielversprechend und liefern eine solide Grundlage für zukünftige klinische Studien. Die Entwicklung dieser Technologie folgt somit einer wegweisenden Strategie im Kampf gegen bislang unheilbare neurodegenerative Erkrankungen.