Die Erforschung des menschlichen Genoms hat in den letzten Jahrzehnten enorme Fortschritte gemacht. Doch trotz der Entschlüsselung der linearen DNA-Sequenz sind viele Fragen zur genauen Regulation der Genaktivität und deren Veränderung bei Krankheiten wie Krebs noch offen. Besonders die räumliche Organisation des Genoms im Zellkern, also seine dreidimensionale (3D) Struktur, gewinnt zunehmend an Bedeutung. Bei primären menschlichen Tumoren liefert das Verständnis dieser komplexen Architektur neue Einsichten für die Krebsforschung und die Entwicklung zielgerichteter Therapien. Das Erbgut eines Menschen besteht aus etwa zwei Metern DNA, die in einem winzigen Zellkern von circa zehn Mikrometern Länge organisiert und gefaltet ist.
Diese Verpackung geschieht keineswegs zufällig, sondern folgt klaren hierarchischen Strukturen, die es ermöglichen, Gene präzise und zeitabhängig zu aktivieren oder zu unterdrücken. Das 3D-Genom bildet dabei eine Grundlage für die Regulation von Genen durch sogenannte cis-regulatorische Elemente, beispielsweise Enhancer, welche teilweise Tausende von Basenpaaren entfernt von den Genen liegen, die sie steuern. In normalen Zellen sind Chromosomen in A- und B-Kompartimente unterteilt. Die A-Kompartimente sind häufiger aktiv und enthalten euchromatische, also entspannte DNA-Regionen, während B-Kompartimente überwiegend heterochromatisch und inaktiv sind. Innerhalb dieser Kompartimente organisieren sich die DNA-Stränge in sogenannten Topologisch Assoziierten Domänen (TADs), welche Interaktionen innerhalb klarer Abgrenzungen ermöglichen, sodass Gene und ihre regulatorischen Elemente in funktionellen Einheiten zusammenarbeiten.
Innerhalb dieser Domänen sind wiederum komplexe Schleifenverbindungen zwischen Enhancern und Promotoren von Genen verankert, die für das präzise Ablesen der Erbinformation entscheidend sind. Krebserkrankungen zeichnen sich durch vielfältige genetische Veränderungen aus. Neben Mutationen in den Kodierenden Sequenzen sind auch Veränderungen in der 3D-Genomstruktur von hoher Relevanz. Diese Veränderungen können durch strukturelle Varianten wie Deletionen, Duplikationen, Inversionen oder Translokationen entstehen. Solche Umordnungen beeinflussen nicht nur direkte DNA-Sequenzen, sondern verändern auch die dreidimensionale Genom-Topologie.
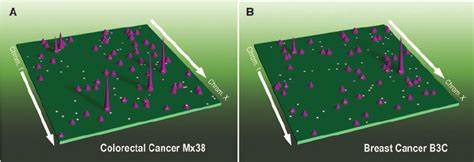
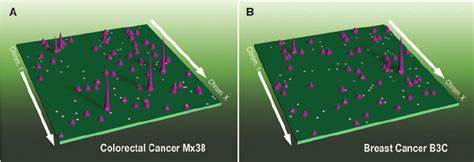
Dadurch können zuvor getrennte regulatorische Bereiche plötzlich zusammenliegen und die Aktivität von Onkogenen – den krebsfördernden Genen – verändern. Ein bedeutender Fortschritt in der Erforschung der 3D-Genomlandschaft menschlicher Tumoren wurde durch die Anwendung der HiChIP-Technologie erzielt. Diese Methode kombiniert chromosome conformation capture mit der spezifischen Anreicherung markanter Histonmodifikationen, beispielsweise der Acetylierung an Lysin 27 des Histon H3 (H3K27ac), die aktive Enhancer kennzeichnet. Anhand von 69 Tumorproben aus 15 verschiedenen Krebsarten aus dem Cancer Genome Atlas (TCGA) wurde die räumliche Architektur der Genome detailliert abgebildet. Dabei konnten über 600.
000 signifikante Interaktionen, insbesondere zwischen Enhancern und Promotoren, identifiziert werden. Die Analyse offenbarte drei archetypische Muster bei der Nutzung von Enhancern in mehr als 100 bekannten Onkogenen: eine statische Nutzung, selektive Verstärkung in bestimmten Krebsarten und eine dynamische Umstrukturierung der Enhancer-Verbindungen. Dieses Wissen eröffnet eine differenzierte Sicht darauf, wie Gene in verschiedenen Tumortypen reguliert und Fehlregulationen bei Krebs entstehen. Besonders beim Myc-Onkogen, einem Schlüsselspieler in vielen Tumorentitäten, zeigte sich eine unterschiedliche Aktivität von Enhancern je nach Gewebe- und Krebsart. So sind in Darmkrebs vor allem regulatorische Elemente auf der 5'-Seite von MYC aktiv, während in Leberkrebs enhancerspezifische Aktivitäten auf der 3'-Seite dominieren.
Diese Varianten der Aktivierung gingen mit unterschiedlichen dreidimensionalen Kontakten im Genom einher, was die Bedeutung der räumlichen Organisation für die Genregulation unterstreicht. Die Auswirkungen genomischer Veränderungen sind jedoch multifaktoriell. Neben Veränderungen in der Enhanceraktivität spielt auch die Kopienzahl von Genen (Copy Number Variation - CNV) eine entscheidende Rolle in der Onkogenexpression. Während bei einigen Genen wie KRAS deutliche Überexpressionen hauptsächlich durch Amplifikationen der Genkopien bedingt sind, kann bei anderen wie MET die veränderte Enhancerinteraktion den dominanten Einfluss darstellen. Diese Individualität in der Genregulation verdeutlicht die Notwendigkeit integrativer Analysen von genomischer Struktur, epigenetischen Markierungen und Transkriptom, um therapeutisch relevante Mechanismen zu verstehen.
Darüber hinaus ist das Tumormikroumfeld, bestehend aus verschiedensten Zelltypen wie Immunzellen, für den Krankheitsverlauf zentral. Die Kombination von HiChIP-Daten mit Einzelzell-ATAC-seq Analysen erlaubt die Zuordnung von spezifischen Enhancer-Promoter-Schleifen zu Zelltypen innerhalb des Tumors. Dabei wurde beispielsweise eine myeloidzellspezifische Enhancer-Promoter-Schleife am PD-L1-Gen, einem wichtigen Immune-Checkpoint, entdeckt. Dies trägt zum besseren Verständnis bei, wie Immunzellen und Tumorzellen das Immunsystem beeinflussen und könnte Implikationen für Immuntherapien haben. Neben Punktmutationen und CNVs nehmen strukturelle Varianten durch genomische Umlagerungen einen besonderen Stellenwert ein.
Solche Veränderungen können neue genetische Kontakte schaffen und dadurch die Genregulation massiv verändern. Besonders die Entstehung von extrachromosomaler DNA (ecDNA), welche als kreisförmige DNAfragmente vorliegt, fördert die massive Überexpression von Onkogenen und lässt sich mit ungünstigen klinischen Verläufen in Zusammenhang bringen. HiChIP-Daten ermöglichten es, durch Integration mit tiefen WGS-Daten diese ecDNA-Komplexitäten zu analysieren und die daraus entstehende umfangreiche Neustrukturierung des Enhancer-Interaktionsnetzwerks zu charakterisieren. Die Ergebnisse aller Untersuchungen weisen darauf hin, dass während die grundlegende Genomkompartimentierung und TAD-Struktur bei Krebs vergleichbar mit gesunden Zellen bleibt, die feingliedrigen Enhancer-Promotor-Beziehungen stark differenzieren und tumorartspezifische Identitäten prägen. Dies unterstreicht die Bedeutung der 3D-Genomforschung in der Präzisionsmedizin.
Durch die Identifizierung von nichtkodierenden Mutationen in enhancerreichen Regionen, die neue Transkriptionsfaktorbindungsstellen schaffen, eröffnen sich weitere Möglichkeiten, bislang unbekannte Krankheitsmechanismen zu erkennen und potenzielle therapeutische Targets zu erschließen. Auch die Erkennung von speziellen chromosomalen Umlagerungen, welche das Genom in neue, funktionelle Schleifen umstrukturieren, kann zukünftig die Klassifikation von Tumoren verbessern und personalisierte Therapieansätze ermöglichen. So können nicht nur die genetischen Kodierungen der Zellen betrachtet werden, sondern auch die 3D-Architektur, welche ein weiterer regulatorischer Layer in der Tumorbiologie ist. Zusammenfassend bietet die Analyse der dreidimensionalen Genomlandschaft primärer menschlicher Krebserkrankungen ein Fenster zur komplexen Welt der Genregulation jenseits der DNA-Sequenz. Die Technologien wie HiChIP in Kombination mit Einzelzelltechniken und tiefem Genomsequenzieren eröffnen neue Wege, molekulare Mechanismen aufzudecken, Therapien zu personalisieren und die Entwicklung von Krebs besser zu verstehen.
Die Erkenntnisse dieser Forschung können langfristig helfen, gezieltere Behandlungsstrategien zu entwickeln, die nicht nur auf Mutationen an sich, sondern auch auf deren epigenetische und räumliche Auswirkungen abzielen – ein vielversprechender Schritt hin zu einer besseren Krebsbekämpfung.