Per- und Polyfluoralkylsubstanzen, kurz PFAS, gehören zu den langlebigsten und umweltrelevantesten Chemikalien des modernen Zeitalters. Sie sind aufgrund ihrer wasser- und fettabweisenden Eigenschaften breit in Industrie, Konsumgütern und vor allem in Aqueous Film-Forming Foams (AFFFs) für Feuerlöschzwecke eingesetzt. Gleichzeitig führen diese Eigenschaften jedoch zu einer erheblichen Umweltbelastung, da PFAS als sogenannte „Forever Chemicals“ bekannt sind, die sich nur schwer abbauen lassen und in Böden, Grundwasser sowie Fauna und Flora anreichern. Aktuelle wissenschaftliche Untersuchungen legen nahe, dass die Selbstassemblierung dieser fluorierten Verbindungen eine zentrale Rolle für ihr Verhalten in der Umwelt spielt und wichtige Hinweise auf ihre Persistenz und Mobilität liefern kann. Die gezielte Erforschung dieses Phänomens eröffnet neue Perspektiven für das Verständnis und letztlich für die Kontrolle sowie Sanierung von PFAS-basierten Kontaminationen.
Unter Selbstassemblierung versteht man die spontane Organisation von Molekülen zu größeren Strukturen durch nicht-kovalente Wechselwirkungen wie hydrophobe Effekte, van-der-Waals-Kräfte oder elektrostatische Anziehung. Fluorosurfactants, die hauptsächlichen Bestandteile vieler PFAS sind, besitzen eine einzigartige Kombination aus hydrophoben fluorierten Ketten und hydrophilen Kopfgruppen. Diese amphiphilen Eigenschaften führen dazu, dass sich PFAS-Moleküle in wässrigen Umgebungen zu supramolekularen Strukturen wie Mizellen, Vesikeln oder Filmen zusammenlagern können. Solche selbstorganisierten Aggregate verändern nicht nur die Löslichkeit und Verteilung der Substanzen, sondern beeinflussen auch deren Transportmechanismen und biologische Verfügbarkeit. Bis vor Kurzem war das Verständnis dieser Selbstassemblierung auf experimentelle Beobachtungen beschränkt, die aufgrund der Komplexität und Chemie der PFAS nur begrenzte Einblicke in molekulare Details liefern konnten.
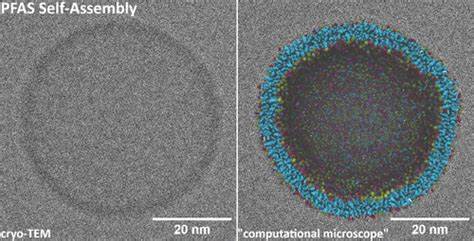
Die neueste Forschung, darunter wegweisende Studien mithilfe sogenannter „computational microscopes“ oder grob gekoppelten molekulardynamischen Simulationen, hat diesem Engpass entgegengewirkt. Durch die Entwicklung spezieller Kraftfeldparameter, etwa im Rahmen des Martini 3 Modells, konnten Wissenschaftler erstmals simulieren, wie sich PFAS-Moleküle unter realistischen Umweltbedingungen zusammenlagern und welche räumlichen und zeitlichen Dynamiken dabei entstehen. Diese computergestützten Simulationen haben gezeigt, dass PFAS je nach chemischer Struktur und Konzentration unterschiedliche supramolekulare Formen annehmen. Die entstandenen Strukturen können von kleinen, kugelförmigen Mizellen bis hin zu komplexen, membranähnlichen Vesikeln reichen, die großen biologischen Kompartimenten ähneln. Solche Aggregate weisen eine erhöhte Stabilität auf, was bedeutet, dass PFAS in der Umwelt länger als zuvor angenommen in gefügten Formen verbleiben können.
Diese Erkenntnis unterstützt die Hypothese, dass sich PFAS nicht nur als einzelne Moleküle, sondern ebenso als großskalige Aggregate im Untergrund ausbreiten und dadurch eine kontinuierliche Quelle für kontaminiertes Wasser darstellen. Die experimentelle Bestätigung dieser Simulationsergebnisse erfolgte durch hochauflösende Kryo-Transmissionselektronenmikroskopie (cryo-TEM). Diese Technik ermöglicht es, die selbstorganisierten Strukturen im nassen Zustand direkt zu visualisieren und die Simulationen empirisch zu validieren. Die übereinstimmenden Befunde aus Computermodellen und Mikroskopie stärken die Aussagekraft der Modelle und eröffnen neue Forschungsansätze zum Monitoring und Management von PFAS-Kontaminationen. Ein weiterer wesentlicher Aspekt der Selbstassemblierung ist ihre dynamische Natur.
Die supramolekularen Aggregate der PFAS sind nicht statisch, sondern unterliegen einem ständigen Gleichgewicht aus Zusammensetzung, Zerfall und Rekonstitution. Dieses Verhalten bestimmt maßgeblich, wie PFAS sich im Boden und Grundwasser verhalten, ob sie an organische Materialen gebunden bleiben oder mobilisiert werden und welche Auswirkungen sie auf biologische Systeme haben. Die Simulationen erlauben es, diese Prozesse auf molekularer Ebene zu verfolgen und somit Vorhersagen über Langzeitverhalten und Sanierungsstrategien zu machen. Die Anwendung der Grobmodellierung mit molekulardynamischen Simulationen ist besonders wertvoll für neue oder wenig erforschte PFAS-Typen. Oft liegen zu diesen Substanzen keine oder nur wenige chemische Standards vor, was experimentelle Untersuchungen erschwert.
Durch die entwickelten computergestützten Modelle lässt sich die Selbstassemblierung und somit auch die Umweltrelevanz theoretisch einschätzen, bevor umfangreiche Laborarbeiten und Umweltstudien durchgeführt werden. Das Verständnis der Selbstorganisationsverhalten von PFAS ist zudem nicht nur für Umweltwissenschaftler von Bedeutung. Es berührt auch die Entwicklung neuer Materialien, die auf Fluorchemie basieren, und hilft dabei, umweltverträglichere Alternativen zu entwickeln. Die Erforschung der Aggregationsdynamik liefert Anhaltspunkte, wie molekulare Modifikationen die Umweltpersistenz verändern können und ermöglicht damit gezielte chemische Innovationen. Abschließend lässt sich sagen, dass die Integration von experimenteller Analytik und fortschrittlichen computergestützten Simulationen einen Paradigmenwechsel in der PFAS-Forschung markiert.
Die gewonnenen Einblicke in die Selbstassemblierung bieten einen tieferen mechanistischen Blick auf das Verhalten dieser langlebigen Schadstoffe und tragen wesentlich dazu bei, sowohl die ökologische Risikobewertung als auch zukünftige technische Lösungsansätze zu verbessern. Der fortlaufende Ausbau dieser Methoden verspricht, das Verständnis der Umweltchemie von PFAS noch präziser zu gestalten und nachhaltige Strategien für den Umgang mit dieser globalen Herausforderung zu entwickeln.