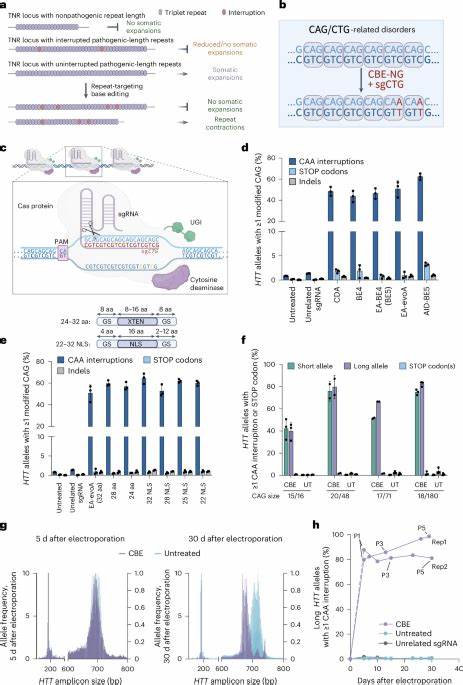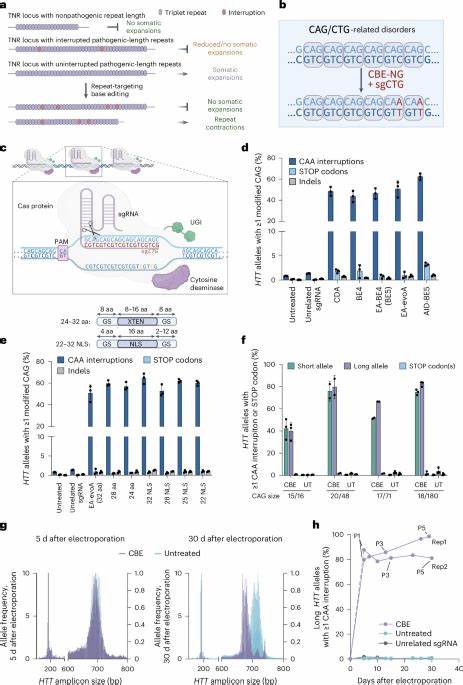
Huntington-Krankheit (HD) gilt als eine der bekanntesten neurodegenerativen Erkrankungen mit genetischer Ursache. Die Krankheit entsteht durch eine pathologische Erweiterung der CAG-Trinukleotid-Wiederholungen im HTT-Gen, welches die Produktion des Huntingtin-Proteins beeinflusst. Mit steigendem Alter und Fortschreiten der Erkrankung neigen betroffen somatische Zellen dazu, diese CAG-Repeatlänge weiter zu erhöhen – ein Prozess, der als somatische Repeat-Expansion bezeichnet wird und wesentlich zur Verschlechterung des klinischen Bildes beiträgt. Die Verhinderung oder Verzögerung dieser Expansion bietet daher einen vielversprechenden therapeutischen Ansatz, um den Ausbruch und Verlauf der Huntington-Krankheit abzumildern oder sogar zu verzögern. Bis vor Kurzem gab es jedoch kaum Möglichkeiten, die molekularen Ursachen der somatischen Instabilität der CAG-Wiederholungen gezielt zu beeinflussen.
Traditionelle Therapieansätze konzentrierten sich meist auf die Symptome oder versuchten, die generelle Expression des Huntingtin-Gens zu senken, was mit erheblichen Nebenwirkungen verbunden sein kann. Der Schlüssel zur gezielten Hemmung der Repeat-Expansion liegt in der genomischen Unterbrechung der CAG-Sequenz, um deren Instabilität zu verringern, ohne die Proteinsequenz selbst entscheidend zu verändern. Hier setzt die innovative Technologie der Basen-Editierung an, die es ermöglicht, gezielt einzelne Nukleotide innerhalb der DNA zu verändern, ohne Doppelstrangbrüche oder größere Mutationen zu verursachen. Forscherteams haben diese Technologie genutzt, um sogenannte CAA-Interruptions—synonyme Codons ähnlich wie CAG, die aber die Instabilität der Wiederholungen deutlich reduzieren—im HTT-Gen einzufügen. Diese natürlichen Unterbrechungen werden mit einer erhöhten Stabilität der CAG-Tracts und einem milderen Krankheitsverlauf in Verbindung gebracht.
Konzepte der Forschung zeigen, dass die künstliche Einführung solcher synonymer Unterbrechungen in den langen CAG-Abschnitten verhindert, dass die Wiederholungen sich in somatischen Zellen weiter ausdehnen. Zahlreiche Experimente an patienteneigenen Fibroblasten haben gezeigt, dass nach Anwendung der Basen-Editoren ein hoher Prozentsatz der Pathogenese-verantwortlichen HTT-Allele genau an den CAG-Wiederholungsstellen mit CAA-Interruptions versehen werden kann. Dabei verbleibt die Funktionalität des Huntingtin-Proteins erhalten, da das CAA-Codon ebenfalls Glutamin kodiert – der essentielle Baustein der repetitive Proteinsequenz. Langzeitbeobachtungen der behandelten Zelllinien verdeutlichen, dass die somatischen Repeat-Expansionen nach der Bearbeitung signifikant eingedämmt werden. Dies bedeutet, dass beschädigte Zellen durch die präzise Genmanipulation eine stabilere genetische Sequenz aufweisen, welche eine forschungsrelevante Basis für therapeutische Interventionen darstellt.
Parallel dazu konnten Untersuchungen bei Knock-in-Mäusen mit humanisiertem HTT-Gen und pathologisch langen CAG-Wiederholungen diese Konzepte bestätigen: Die Einbringung von CAA-Interruptions durch Basen-Editoren in vivo führte zu einer messbaren Abnahme der somatischen Erweiterungen in besonders betroffenen Hirnregionen wie Cortex und Striatum. Die Umsetzung der Gen-Bearbeitung erfolgt häufig über Adeno-assoziierte Viren (AAV), die die Basen-Editor-Komponenten effizient in Zielneuronen einbringen können. Neonatale intraventrikuläre Injektionen mit speziell optimierten AAV9-Serotypen gewährleisten eine erfolgreiche Expression der Base Editing-Systeme in kritischen Hirnarealen. Die genetischen Veränderungen zeigen sich dauerhaft und führen zu einer anhaltenden Reduktion der Repeat-Expansion, wodurch die Wahrscheinlichkeit eines schnellen Fortschreitens der Erkrankung bezüglich Zelltoxizität und neurodegenerativen Prozessen deutlich minimiert werden kann. Die präzise Bearbeitung der trinukleotidischen Repeats ist dabei nicht nur wirksam, sondern auch mit einem günstigen Sicherheitsprofil verbunden.
Die Off-Target-Analysen zeigen, dass die Veränderungen überwiegend auf die Zielsequenzen beschränkt bleiben und keine signifikanten Nebenwirkungen auf andere Kodierungs- oder regulatorische Genabschnitte befürchtet werden müssen. Die eingeschleusten Interruptions-Codons sind dabei ähnlich natürlich vorkommenden Varianten und führen vorrangig zu synonymen, also proteinneutralen, Änderungen. Diese Fortschritte eröffnen neue Perspektiven in der Huntington-Therapie. Die langfristige Stabilisierung der genetischen Repeat-Tracts könnte den Krankheitsbeginn verzögern oder sogar aufhalten. Gleichzeitig wird dadurch die Grundlage geschaffen, die molekularen Treiber des Krankheitsverlaufs direkt zu adressieren, anstatt nur die Symptome zu behandeln.
Die Kombination von präziser Genomeditierung mit etablierten oder zukünftigen Therapieformen verspricht, die Lebensqualität der Patienten massiv zu verbessern. Neben Huntington ist das Konzept der Gen-Stabilisierung durch Interruptionsinsertionen auch für andere Trinukleotid-Repeat-Erkrankungen relevant. Ähnliche Mechanismen liegen etwa der Friedreich-Ataxie zugrunde, bei der GAA-Repeats im FXN-Gen ebenfalls instabil sind. Auch hier konnten Basen-Editoren eingesetzt werden, um Interruptionssequenzen einzufügen, sodass sich therapeutische Strategien für eine Reihe von neurodegenerativen Erkrankungen ableiten lassen. Wichtig ist die Erkenntnis, dass die somatische Instabilität der Repeats ein entscheidender Faktor für den Krankheitsverlauf ist, der bisher kaum adressiert wurde.