Huntington-Krankheit (HD) ist eine verheerende neurodegenerative Erkrankung, die durch eine pathologische Expansion der CAG-Trinukleotid-Wiederholung im Huntingtin-Gen (HTT) verursacht wird. Diese Wiederholungen kodieren für eine Polyglutamin-Sequenz, deren Länge eng mit dem Krankheitsverlauf korreliert. Je länger die ununterbrochene Wiederholungsstrecke, desto früher liegt in der Regel der Krankheitsbeginn, und desto schwerwiegender ist der Verlauf. Die Instabilität und Ausdehnung dieser CAG-Trinukleotide spielt eine zentrale Rolle bei der Pathogenese und hat sich als vielversprechender Zielpunkt für therapeutische Interventionen erwiesen. Jüngste Fortschritte in der gezielten Genomeditierung bieten Hoffnung auf eine innovative Behandlung, die das Fortschreiten der Krankheit auf molekularer Ebene verlangsamen oder gar stoppen könnte.
Somatische Repeat-Expansionen und ihre Rolle bei Huntington Die somatische Instabilität der CAG-Wiederholungen bedeutet, dass sich die Länge der Wiederholungssequenzen im Verlauf des Lebens in einzelnen Körperzellen zunehmend verändern kann. Besonders ausgeprägt ist diese Expansion in Geweben des zentralen Nervensystems, vor allem in den für Huntington relevanten Hirnbereichen, wie dem Striatum und der Großhirnrinde. Diese Erweiterungen können neuronale Funktion und Überleben beeinträchtigen und beschleunigen somit den Krankheitsverlauf. Wesentliche Studien haben gezeigt, dass nicht die Ausgangslänge der CAG-Repeats allein, sondern vor allem deren somatische Instabilität zum Auftreten und zur Progression der Krankheit beiträgt. Die Ursprünge der somatischen Expansion liegen in molekularen Mechanismen wie der DNA-Reparatur, insbesondere bei fehlgeleiteten Reparaturprozessen, die zu ungleichmäßiger Verlängerung der Trinukleotidsequenzen führen.
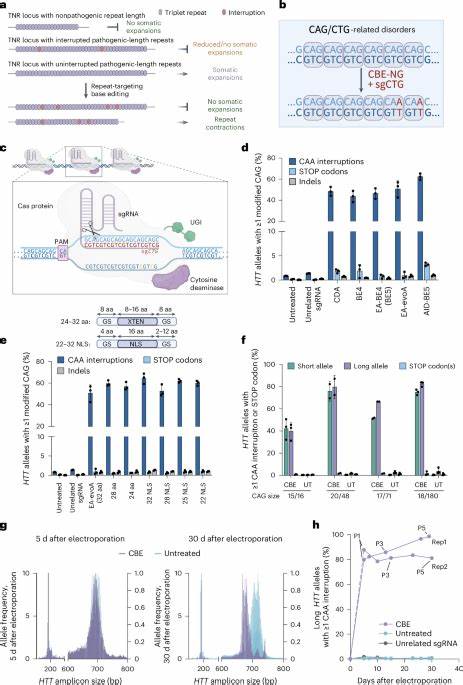
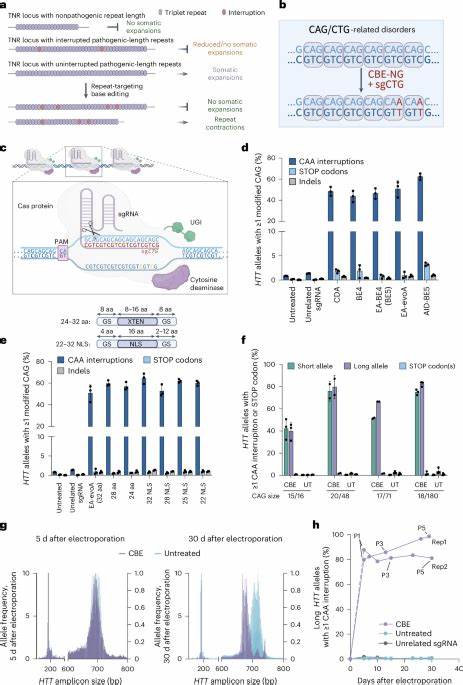
Zudem fördern komplexe DNA-Strukturen, wie Haarpinne oder Schleifen innerhalb der CAG-Trinukleotide, diese Instabilität. Das Verstehen dieser Mechanismen ist für die Entwicklung von Therapien zur Stabilisierung oder Korrektur der Repeat-Länge essenziell. Natürlich vorkommende Interruptionsmutationen als Vorbild In der Population existieren allelische Varianten, bei denen innerhalb der langen CAG-Sequenzen einzelne Synonyme (z. B. CAA) als Interruptionsmotive auftreten.
Diese genetischen Unterbrechungen wirken stabilisierend auf die Wiederholungsregion und sind mit einem späteren Krankheitsbeginn sowie einer verminderten somatischen Expansion assoziiert. Studien haben gezeigt, dass ein Austausch einzelner Codons von CAG zu CAA, die beide für die Aminosäure Glutamin kodieren, die Bildung von instabilen DNA-Strukturen reduziert und so die genetische Stabilität erhöht. Das Phänomen solcher natürlicher Interruptionsmutationen ist nicht nur auf Huntington beschränkt, sondern findet sich auch bei anderen Trinukleotid-Expansionserkrankungen wie der Friedreich-Ataxie. Ihr Vorbild dient als Inspiration für therapeutische Ansätze, die diese Interruptionsmotive gezielt in pathogene Repeat-Sequenzen einbringen, um die schädliche Expansion zu verhindern. Moderne Präzisionsgenom-Editing-Technologien ermöglichen gezielte Veränderungen Die Entwicklung der Base Editing-Technologie revolutioniert die Möglichkeiten der gezielten DNA-Modifikation.
Im Unterschied zu klassischen CRISPR/Cas9-Systemen, die Doppelstrangbrüche induzieren, ermöglichen Cytosin- (CBE) und Adenin-Base-Editoren (ABE) die direkte und präzise Änderung einzelner Nukleotide ohne DNA-Strangbruch. Für Huntington ist vor allem die Verwendung von CBEs relevant, da sie C•G zu T•A Substitutionen gezielt einfügen können, etwa um CAG zu CAA zu modifizieren. Jüngste Forschungen zeigen, dass durch sgRNAs, die die CAG-Sequenzen direkt adressieren, Base Editor-Systeme in Patientenfibroblasten und murinen Modellen effizient Interruptionsmutationen in den HTT-Genen erzeugen. Diese Mutationen sind synonym, verändern also nicht die Aminosäuresequenz, verhindern aber die Bildung schädlicher DNA-Strukturen und minimieren die somatische Expansion. Diese Ergebnisse wurden anhand modernster molekularbiologischer Techniken wie Hochdurchsatzsequenzierung und Fragmentanalyse bestätigt.
Wissenschaftler konnten zeigen, dass Patientenfibroblasten nach Base Editing im Labor eine deutlich geringere Tendenz zur somatischen Repeat-Expansion über Zellpassagen aufweisen. In vivo Experimente mit Htt.Q111 Mäusen, einem etablierten HD-Modell, wurden durch adenoassoziierte Virusvektoren (AAV9) in Neugeborenen mittels intrazerebroventrikulärer Injektion mit Base Editor-Machinery behandelt. Die Ergebnisse zeigten signifikante Reduktionen der somatischen Repeat-Expansion in Gehirnregionen relevant für HD, insbesondere in Cortex und Striatum. Auswirkungen auf die Krankheitsprogression und therapeutische Potenziale Die Reduktion der somatischen Expansion ist von enormem klinischem Interesse, da sie potenziell den Zeitpunkt des Krankheitsbeginns verzögern oder die Symptomatik abschwächen könnte.
Studien zeigen, dass der Zeitpunkt der Ausprägung sich mit einer einzigen Unterbrechung in der CAG-Strecke um mehr als ein Jahrzehnt verschieben lässt. Somit könnte ein Therapieansatz, der Interruptionsmutationen gezielt einführt, neben der veränderten DNA auch funktionelle posititve Effekte auf molekularer Ebene bewirken. Dabei beinhaltet der Vorteil der Base Editing-Technologie, neben der hohen Zielgenauigkeit, auch die Möglichkeit, Veränderungen ohne dauerhafte DNA-Doppelstrangbrüche zu bewirken, was potenziell das Risiko unerwünschter Mutationen oder zellulärer Toxizität minimiert. Die AAV-vermittelte Übertragung in postnatale Tiere zeigt, dass eine langanhaltende Expression der Editoren in neuronalen Zellen möglich und wirksam ist. Herausforderungen hinsichtlich Off-Target-Effekten und Sicherheit Trotz vielversprechender Ergebnisse existieren wichtige Sicherheitsfragen.
Die Base Editor können auch an ähnlichen Sequenzen unerwünschte Änderungen (Off-Target-Editing) verursachen. Umfangreiche Genomanalysen zeigen jedoch, dass die meisten Off-Target-Mutationen in nichtkodierenden Regionen auftreten und viele editierte Varianten bereits in der menschlichen Bevölkerung vorhanden sind oder keine schädlichen Proteinveränderungen hervorrufen. Nichtdestotrotz wird die Minimierung von Off-Target-Effekten als essentiell angesehen. Fortschritte in der Protein-Engineering-Technologie haben zu Base Editor Varianten mit verbesserter Spezifität geführt. Zudem wird die Entwicklung alternativer Liefermethoden erforscht, um eine gezielte und kontrollierte Expression der Editoren in relevanten Zielzellen zu gewährleisten, die mögliche Risiken weiter reduzieren können.
Zukunftsperspektiven und klinische Translation Das Editieren von CAG-Repeat-Regionen im HTT-Gen stellt einen revolutionären Ansatz zur Behandlung von Huntington dar. Ausgangspunkt zukünftiger Anwendung ist die kontinuierliche Verbesserung der Effizienz sowie der Sicherheit der Base Editing-Technologie und ihrer Abgabe in den Patienten. Neben der Behandlung von Huntington könnte ein ähnlicher Ansatz auch bei anderen Trinukleotid-Erkrankungen wie Friedreich-Ataxie oder spinocerebellären Ataxien Anwendung finden, da auch dort der Einfluss der Repeat-Instabilität entscheidend ist. Die Anpassung an unterschiedliche Repeattypen und molekulare Gegebenheiten eröffnet Chancen für eine breite therapeutische Anwendung. Auswahl geeigneter Tiermodelle, längerfristige Studien sowie die Entwicklung gezielter Enzymvarianten sind wichtige Schritte, die eine klinische Erprobung vorbereiten.