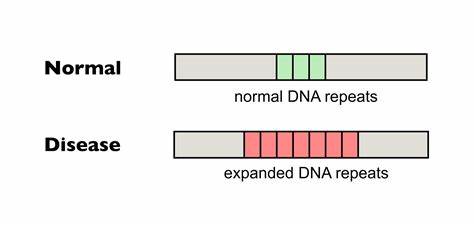
Huntington ist eine genetische Erkrankung, die durch die Erweiterung einer bestimmten trinukleotidischen Wiederholung – den CAG-Repeat – im Huntingtin-Gen ausgelöst wird. Diese pathologische Expansion führt zu einer fortschreitenden neurodegenerativen Erkrankung, die mit motorischen Störungen, kognitivem Abbau und psychiatrischen Symptomen einhergeht. Seit ihrer Entdeckung stellt die instabile Natur dieser repetitiven DNA-Abschnitte eine große Herausforderung für Wissenschaftler und Mediziner dar. Diese Instabilität, die in verschiedenen Körperzellen auftreten kann, führt im Verlauf des Lebens eines Patienten zu einer somatischen Repeat-Expansion – einer weiteren Verlängerung der Wiederholungen in bestimmten Geweben, vor allem im Gehirn. Die somatische Expansion korreliert eng mit dem Erkrankungsverlauf und dem Schweregrad der Symptome.
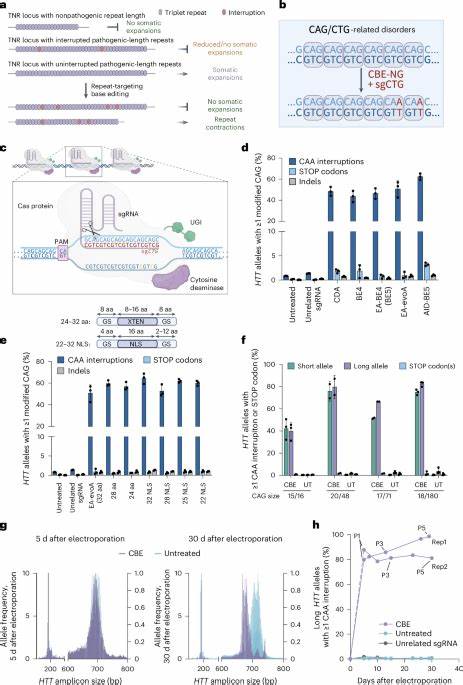
Ein signifikantes therapeutisches Potenzial liegt daher in der Hemmung oder Umkehrung dieser somatischen Repeat-Expansionsprozesse. Neuentwickelte Gen-Editing-Technologien, insbesondere die sogenannten Base-Editing-Verfahren, bieten vielversprechende Möglichkeiten, das genbasierte Grundproblem der CAG-Repeat-Erweiterung gezielt anzugehen. Dabei ermöglichen Cytosin- oder Adenin-Base-Editoren die Punktmutation einzelner Nukleotide ohne den kompletten Durchtrennung der DNA-Stränge, was eine besonders präzise und nebenwirkungsarme Möglichkeit bietet. Forscher haben entdeckt, dass bestimmte unterbrechende Nukleotidsequenzen – sogenannte Interruptions – innerhalb der CAG-Repeats, insbesondere Synonyme Veränderungen wie CAA-Stellen im Repeat-Trakt, die Stabilität der Sequenz erhöhen und die Neigung zur Expansion reduzieren können. Dieses natürliche Phänomen haben Wissenschaftler zum Anlass genommen, mittels Base-Editing gezielt solche Interruptions künstlich in pathogene CAG-Repeat-Trakte einzufügen.
Ziel ist es, die molekulare Struktur der DNA so zu verändern, dass sie weniger anfällig für Faltung und Fehlreparaturen wird, die letztlich die Expansion fördern. In Laboruntersuchungen an Huntington-Patientenzellen wurde die Wirksamkeit dieser Methode bereits beeindruckend unter Beweis gestellt. Durch Elektroporation von mRNA-codierten Base-Editoren und passenden Leitstrang-RNAs (sgRNAs), die auf die CAG-Trakte abzielen, konnten in einem hohen Anteil der Zellen CAA-Interruptions effizient und langlebig eingebaut werden. Bemerkenswert ist dabei, dass die Effizienz der Bearbeitung an längeren, also pathologischen, Allelen höher ausfällt, was die Selektivität und Effektivität dieser Methode unterstreicht. Langfristige Beobachtungen zeigten, dass die somatische Expansion nach Base-Editing-Behandlung signifikant reduziert oder sogar teilweise rückgängig gemacht wurde.
Erkrankungsverlauf prognostisch negative CAG-Verlängerungen konnten demnach durch gezielte Interruptionsverteilung vermieden werden, was einen entscheidenden Fortschritt gegenüber früheren Ansätzen darstellt. Die Anwendung dieser Präzisionsgenom-Editing-Techniken ist über reine Zellkulturen hinausgegangen. In vivo Experimente an genetisch veränderten Mausmodellen für Huntington zeigten, dass die intrazerebroventrikuläre Verabreichung von von Adeno-assoziierten Viren (AAV9) kodierten Base-Editoren zu einer substantiellen und zielgerichteten Bearbeitung der CAG-Repeat-Sequenz in kortikalen und striatalen Neuronen führt. Die entsprechenden Interruptions konnten über Wochen bis Monate eingeführt und in den behandelten Hirnregionen nachgewiesen werden. Dadurch wurde die üblicherweise beobachtete somatische Repeat-Expansion deutlich gedämpft, was die Schutzwirkung und den therapeutischen Nutzen der Interruptions bestätigt.
Auch wenn sich die Modelle bisher noch nicht zu vollwertigen Krankheitsbehandlungsstudien weiterentwickelt haben, bilden diese Erkenntnisse eine solide Grundlage für zukünftige Therapieansätze. Die Herausforderung bei der Genom-Editing-Therapie liegt nicht nur in der Effizienz, sondern gerade auch in der Sicherheit der eingesetzten Werkzeuge. Base-Editing-Methoden bringen zwar eine hohe Zielgenauigkeit mit sich, können jedoch potenziell auch Nebenwirkungen auslösen – insbesondere durch Off-Target-Effekte an anderen genomischen Stellen. Aus diesem Grund wurde bei den Huntington-spezifischen Experimenten ein umfangreiches Screening und Monitoring der Off-Target-Editing-Aktivitäten durchgeführt. Dieses umfasste sowohl computergestützte Vorhersagen als auch experimentelle Nachweismethoden wie CIRCLE-seq und Hochdurchsatz-WGS-Analysen.
Alles in allem zeigten sich nur wenige und überwiegend harmlose Off-Target-Mutationen, die größtenteils nicht zu Veränderungen der Proteinsequenzen führten oder natürliche Varianten im menschlichen Genom widerspiegelten. Das Verhältnis von Nutzen zu Risiko ist daher vielversprechend, wenngleich weitere Studien zur Langzeit-Sicherheitsbewertung zwingend erforderlich sind. Parallel zu Huntington wurde ein ähnliches Prinzip bei Friedreich-Ataxie erforscht, einer Erkrankung, die durch die Expansion von GAA-Trinukleotiden in einem anderen Gen verursacht wird. Auch hier konnten durch Adenin-Base-Editing Interruptions künstlich eingeführt und somit die somatische Expansion signifikant gemindert werden. Diese Parallele verdeutlicht den universellen Wert des Base-Editing-Ansatzes für eine ganze Klasse von Trinukleotid-Repeat-Erkrankungen und untermauert die Bedeutung weiterer Investitionen in diese Technologie.
Die therapeutischen Perspektiven für Patienten mit Huntington unterstreichen nicht nur die Bedeutung von Gen-Editing-Anwendungen zur Verhinderung weiterer Pathologie, sondern auch die Relevanz früher oder präventiver Eingriffe, um das Fortschreiten zu verzögern oder gar aufzuhalten. Die Tatsache, dass somatische Repeat-Expansionen meist über Jahrzehnte erfolgen und erst lange nach Krankheitsbeginn klinisch relevant werden, eröffnet ein wertvolles Zeitfenster für Therapie. Mit gezieltem Base-Editing kann möglicherweise priorisiert werden, die Instabilität der CAG-Repets präventiv zu stabilisieren, sodass die kritische Schwelle für den Krankheitsausbruch nicht überschritten wird. Die Forschung auf diesem Gebiet schreitet rasch voran. Die Optimierung der Base-Editoren, ihre effizientere und sicherere Proteinstruktur und die Entwicklung besserer Vektor-Systeme für die zielgerichtete Gentherapie im Gehirn zählen zu den aktuellen Prioritäten.
Daneben wird intensiv an alternativen Editierstrategien wie Prime-Editing gearbeitet, die noch subtilere und komplexere DNA-Modifikationen ermöglichen. Darüber hinaus gewinnen nicht-virale Lieferformen und kurzzeitige expressionsbasierte Ansätze an Bedeutung, die klinisch flexiblere und nebenwirkungsarme Behandlungsoptionen versprechen. Abschließend lässt sich festhalten, dass die gezielte Bearbeitung pathogener CAG-Repeats durch Base-Editing einen revolutionären Schritt in der Huntington-Forschung darstellt. Indem sie somatische Repeat-Expansionen verringert und damit den genetischen Grundmechanismus der Erkrankung direkt adressiert, eröffnet diese Technologie neue Hoffnung auf wirksame Therapien. Zwar stehen klinische Anwendungen noch am Anfang, jedoch könnten die publizierten Studienergebnisse ein Fundament für zukünftige Interventionen bilden, die das Leben von Patienten maßgeblich verbessern.
Darüber hinaus bietet dieser Ansatz eine Blaupause für den Umgang mit anderen Repeat-Expansion-Erkrankungen, was das klinische Potenzial nochmals erheblich steigert. Auf dem Weg zur Anwendung im Menschen sind jedoch noch Fragen zu klären, etwa zur optimalen Dosis, Dauer der Basen-Editor-Expression, langfristigen Sicherheit und der volleffektiven Distribution im menschlichen Gehirn und anderen betroffenen Organsystemen. Die Kombination interdisziplinärer Expertise aus Molekulargenetik, Neurologie, Bioinformatik und klinischer Forschung wird entscheidend sein, um diese Herausforderungen zu meistern und das Potenzial der Genom-Editing-Technologien bei Huntington und verwandten Erkrankungen voll auszuschöpfen.