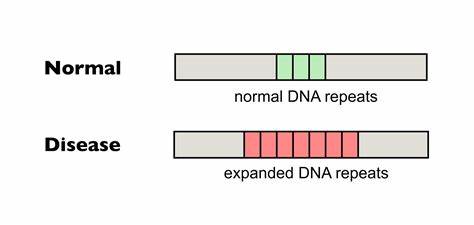
Die Huntington-Krankheit zählt zu den bislang unheilbaren neurodegenerativen Erkrankungen und wird durch die Expansion spezifischer Gen-Repeats, insbesondere der CAG-Tripletts im Huntingtin-Gen (HTT), verursacht. Die Länge dieser sogenannten Trinukleotidwiederholungen bestimmt wesentlich den Ausbruch und Schweregrad der Erkrankung. Eine charakteristische Eigenschaft ist die somatische Instabilität dieser Sequenzen, die sich im Laufe des Lebens in bestimmten Zelltypen kontinuierlich verlängern können und dadurch die zelluläre Funktion zunehmend beeinträchtigen. Jüngste wissenschaftliche Durchbrüche nutzen innovative Basen-Editing-Technologien, um diese krankheitsrelevanten Repeats direkt zu bearbeiten, was zu einer signifikanten Reduktion der somatischen Expansionen in den Zellen von Patienten führt und somit neue Hoffnung für Therapien bietet. Die pathologische CAG-Expansion im HTT-Gen führt zur Bildung eines mutierten Huntingtin-Proteins mit verlängerten Polyglutamin-(PolyQ)-Abschnitten, deren toxische Wirkung zu neuronalem Absterben und den für Huntington typischen Symptomen wie Bewegungsstörungen, kognitivem Verfall und psychischen Veränderungen führt.
Die Repeatlänge ist dabei nicht statisch, sondern unterliegt einer dynamischen Instabilität, die nicht nur in der Keimbahn, sondern auch in somatischen Zellen auftritt. Untersuchungen haben gezeigt, dass besonders im zentralen Nervensystem, etwa in Striatum und Kortex, die Repeatlängen im Laufe des Lebens durch somatische Expansionen zunehmen – dieser Prozess trägt maßgeblich zur Krankheitsprogression bei. Die Herausforderung besteht darin, diese instabilen Repeats gezielt und schonend so zu modifizieren, dass ihre Ausdehnung verhindert oder sogar rückgängig gemacht wird, ohne die DNA an anderer Stelle zu schädigen. Hier kommen cytosin- und adeninbasierte Baseneditoren zum Einsatz, die es erlauben, einzelne Nukleotidpaare umzuwandeln, ohne Doppelstrangbrüche zu induzieren, was das Risiko unerwünschter Effekte erheblich verringert. Der innovative Ansatz beruht auf der gezielten Einführung von sogenannten Unterbrechungen in die CAG-Repeats, indem einzelne Cytosin-zu-Thymin-(C>T)- oder Adenin-zu-Guanin-(A>G)-Übergänge eingebaut werden.
Diese einzelnen Nukleotidsubstitutionen erzeugen im Endeffekt geringfügige Abweichungen im Repeatsystem, die jedoch klinisch signifikant sind. Untersuchungen zeigten, dass natürliche Unterbrechungen, wie die Umwandlung von CAG zu CAA (beide codieren für Glutamin), die somatische Instabilität vermindern und den Ausbruch der Krankheit verzögern können. Mithilfe von Baseneditoren wird dieser natürliche Mechanismus therapeutisch nachgeahmt. Studien am Beispiel humaner Fibroblasten von Huntington-Patienten mit langen CAG-Expansions belegen, dass die Anwendung von cytosinbasiertem Editing zu einer substanziellen Unterbrechung der Repeats führt. Die behandelten Zellen zeigten signifikant weniger somatische Expansionen über mehrere Zellteilungen hinweg verglichen mit unbehandelten Kontrollen.
Diese Befunde markieren einen entscheidenden Schritt in der Entwicklung effektiver Gentherapien, die auf die Ursache der Huntington Erkrankung zielen. Weiterhin ermöglicht der Einsatz von Adenin-Baseneditoren die Modifikation anderer schädlicher Repeats, wie die GAA-Expansionen im FXN-Gen bei Friedreich-Ataxie, ebenfalls eine TNR-Erkrankung mit ähnlicher somatischer Instabilität. Auch hier konnten in vitro und in vivo gezielte Unterbrechungen eingefügt werden, die eine Verlangsamung oder Begrenzung der Repeat-Expansionen bewirken. Die Anwendung dieser Technologien in vivo erfolgte in Mausmodellen mit humanisierten HTT- oder FXN-Genen, die die somatische Instabilität der Repeats simulieren. Durch die Verwendung von Adeno-assoziierten Viren (AAV9) als Vektor für die Baseneditor-Gene konnte eine effiziente Transduktion von Gehirngeweben wie Kortex und Striatum erreicht werden.
Die Ergebnisse zeigten neben stabiler und anhaltender Editierung der Repeats eine signifikante Verkleinerung der Wiederholungen durch verminderte Expansion und erhöhte Kontraktionen. Dies signalisiert eine vielversprechende therapeutische Möglichkeit, die somatische Repeat-Expansion zu kontrollieren und so die Krankheitsprogression zu verlangsamen. Neben der Effizienz wurde bei diesen Studien auch ein umfangreiches Off-Target-Screening durchgeführt. Hierbei zeigte sich, dass die Baseneditoren trotz der hohen Anzahl ähnlicher Repeat-Sequenzen im Genom hauptsächlich an den zielgerichteten Loci wirkten und wenige unerwünschte Mutationen in protein-kodierenden Regionen induzierten. Die meisten Nebenwirkungen bestanden aus ungefährlichen synonymer Mutationen oder üblichen natürlichen Genvarianten.
Trotzdem ist die mögliche langfristige Wirkung dieser Veränderungen Gegenstand weiterer Forschung, um die Sicherheit eines klinischen Einsatzes sicherzustellen. Der therapeutische Wert der Bearbeitung von CAG-Repeats bei Huntington beruht auf der Reduktion der instabilen Repeatlänge vor dem kritischen Schwellenwert, der den neuronalen Zerfall beschleunigt. Studien zeigen, dass das Einbringen von Unterbrechungen innerhalb der Repeat-Trakte nicht nur die Expansion hemmt, sondern auch Wiederholungen verkürzen kann, wodurch zelluläre Funktionen potentiell erhalten bleiben und die neurodegenerativen Prozesse verlangsamt werden. Diese Fortschritte bieten eine neue Perspektive im Kampf gegen TNR-bedingte Krankheiten, die bislang primär symptomatisch behandelt wurden. Die Möglichkeit, die genetische Ursache präzise zu adressieren, stellt einen Paradigmenwechsel dar und könnte langfristig das klinische Management von Huntington deutlich verändern.
Voraussetzung für den Erfolg bleibt jedoch die weitere Optimierung der Liefersysteme, um eine effiziente und breitflächige Verteilung im menschlichen Gehirn zu gewährleisten, sowie die Minimierung von immunologischen Reaktionen und Off-Target-Effekten. Außerdem sind aussagekräftige Langzeitstudien in Tiermodellen und später klinische Studien notwendig, um das Potential und die Sicherheit dieser Ansätze abschließend zu bewerten. Zusammenfassend lässt sich sagen, dass das gezielte Editing der CAG-Repeats im Huntingtin-Gen und die damit verbundene Reduktion somatischer Repeat-Expansionen eine vielversprechende innovative Strategie zur Behandlung der Huntington-Krankheit darstellt. Durch den Einsatz präziser Baseneditoren können natürliche genetische Variationen mimetisiert werden, welche die Instabilität der Repeats mindern. Dieser bahnbrechende Ansatz nicht nur die molekulare Grundlage der Krankheit adressieren, sondern auch das Fortschreiten der Neurodegeneration verzögern oder stoppen, wodurch Patienten bessere therapeutische Optionen geboten werden könnten.
Die kontinuierliche Forschung und technologische Weiterentwicklung sowie die sorgfältige Bewertung von Sicherheit und Effektivität werden maßgeblich über den zukünftigen klinischen Einsatz entscheiden.





![The Talent Stack [video]](/images/D34EDCCA-5B71-4217-995D-6395D79180BB)