Die Huntington-Krankheit ist eine verheerende genetische Erkrankung, die durch die Pathogenese langgestreckter CAG-Trinukleotid-Repeat-Expansions im Huntingtin-Gen (HTT) verursacht wird. Diese sich ausdehnenden Wiederholungen führen zur Bildung toxischer Proteine und zur fortschreitenden neurodegenerativen Schädigung, die sich meist im mittleren Erwachsenenalter manifestiert. Trotz Jahrzehnten intensiver Forschung existiert bislang keine kurative Therapie, die das Fortschreiten dieser Erkrankung wirksam aufhält oder gar umkehrt. Die Suche nach innovativen Ansätzen zur Stabilisierung und Modifikation der genetischen Ursache ist daher von größter Bedeutung. In den letzten Jahren hat das sogenannte Basen-Editing als neue präzise Methode der Genom-Editierung zunehmend Aufmerksamkeit erhalten.
Anders als traditionelle CRISPR-Cas9-Technologien, die DNA-Doppelstrangbrüche induzieren, ermöglichen Basen-Editoren die gezielte, enzymatisch vermittelte Änderung einzelner Basenpaare ohne dabei die DNA-Strangkontinuität zu zerstören. Diese Methode ist besonders vielversprechend für die gezielte Modifikation repetitiver DNA-Sequenzen, wie sie bei Trinukleotid-Repeat-Erkrankungen auftreten. Forscher konnten nun zeigen, dass durch die Einführung von sogenannten 'interruptions' – also gezielten Basenveränderungen innerhalb der lange ausgedehnten CAG-Repeat-Regionen des HTT-Gens – die somatische Instabilität dieser Repeat-Abschnitte vermindert wird. Diese Unterbrechungen sind in derartigen DNA-Trinukleotid-Regionen natürlicherweise bekannt und mit einem verzögerten Krankheitseintritt in Verbindung gebracht worden. Die gezielte Umsetzung dieser positiven genetischen Variationen mithilfe von Basen-Editoren steht somit im Zentrum aktueller therapeutischer Innovationen.
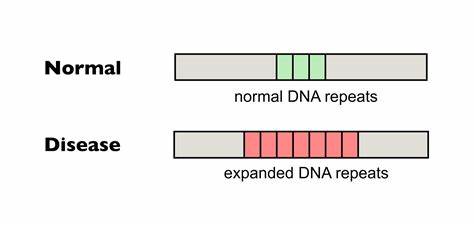
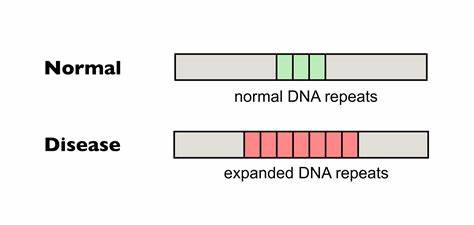
Bei der Huntington-Krankheit ist vor allem die Erweiterung von CAG-Trinukleotid-Wiederholungen im Exon 1 des Huntingtin-Gens verantwortlich für die schädlichen zellulären Effekte. Wenn eine kritische Länge überschritten wird, neigen diese Repeat-Abschnitte dazu, sich in bestimmten somatischen Zellen weiter auszudehnen, was zu einer Verstärkung der pathogenen Proteine und verstärkten neurodegenerativen Schäden führt. Die Länge der Repeat-Tracts bei Geburt bestimmt weitgehend das Erkrankungsalter und den Schweregrad, aber die im Laufe des Lebens auftretenden somatischen Expansionen können die klinische Manifestation weiter verschlimmern. In Zellkulturen aus Huntington-Patienten wurde inzwischen belegt, dass die gezielte Modifikation der CAG-Repeats durch Cytosin-Basen-Editoren, welche die Basenfolge subtil in etwaige CAA-Codons umwandeln, eine Form von synonymer Interruption erzeugen, die keine Aminosäureänderung im Protein verursacht. Diese CAA-Interruptionsvorgänge verhindern die somatische Ausdehnung der Repeat-Regionen in betroffenen Zellen und fördern teilweise sogar eine leichte Kontraktion der CAG-Tracts.
Diese Stabilisierung der DNA-Sequenz ist für den Krankheitsverlauf entscheidend, da sie das Fortschreiten der neuronalen Schädigung verlangsamen könnte. Die Anwendung dieser präzisen lokalen Basenveränderung erfolgte unter Verwendung optimierter Cas9-Varianten mit spezifischer Guide-RNA, die auf die wiederkehrenden CTG-Repeats auf dem Gegenstrang des HTT-Gens abzielen. Mit Hilfe dieser Technik gelang es, in primären Fibroblasten von Huntington-Patienten einen hohen Anteil an bearbeiteten HTT-Allelen zu erzielen – in manchen Fällen mehr als zwei Drittel der Allele zeigten zumindest eine einzige Unterbrechung im ansonsten homogenen CAG-Repeat-Stretch. Diese Interruptionsmuster korrelierten mit einer signifikanten Abnahme der somatischen Expansion in vitro, was höchsten klinischen Erwartungen entspricht. Der enorme Fortschritt bei der Wirksamkeit dieser Methode wurde zusätzlich durch Anwendung in vivo in Mausmodellen belegt.
Insbesondere eine humane Huntingtin-Gen-KI-Mauslinie (Htt.Q111) zeigte nach adenoassoziierter Virus (AAV)-Vektor-vermittelter Expression der Basen-Editoren im Zentralnervensystem eine deutliche Reduktion der somatischen CAG-Repeat-Expansion in Gehirnregionen wie dem Kortex und dem Striatum. Dies ist besonders bedeutsam, da gerade diese Regionen bei Huntington besonders anfällig für neurodegenerative Effekte sind. Es zeigte sich, dass die Basiseditoren nicht nur die Expansion verhinderten, sondern auch eine moderate Kontraktion der vorhandenen Repeat-Längen induzierten. Damit bietet das Verfahren nicht nur eine präventive, sondern auch eine therapeutische Möglichkeit bei bereits manifestierter Mutation an.
In den behandelten Tieren konnten bis zu sechs oder mehr Interruptionscodons im HTT-Gen nachgewiesen werden, was die hohe Effizienz der Editorsysteme verdeutlicht. Von großer Bedeutung für vergleichbare therapeutische Ansätze sind mögliche unerwünschte Veränderungen im Genom außerhalb des Zielgenbereichs. Umfangreiche Untersuchungen mittels CIRCLE-seq und hochauflösender Ganzgenomsequenzierung verdeutlichten, dass die Off-Target-Aktivitäten der verwendeten Basen-Editoren hauptsächlich an genomischen Regionen mit hoher Ähnlichkeit zu den gezielten Repeat-Sequenzen lokalisiert sind. Dort kommt es überwiegend zu synonymer Codonveränderung oder zu benignen Varianten, die nicht mit pathogenen Effekten assoziiert sind. Nonsynonyme Mutationen traten nur selten auf und meisten dieser Veränderungen wurden computergestützt als wahrscheinlich benign eingeschätzt.
Die sorgfältige Auswahl entsprechender Cas9-Varianten mit erweiterten PAM-Erkennungssequenzen verbesserte zudem die Spezifität der Bindung an die Zielregionen. Die Erkenntnisse beleuchten einen ganz zentralen Aspekt der Huntington-Forschung: Die genomische Repeat-Instabilität ist ein dynamischer Faktor, dessen Modulation herzlich zur Pathogenese der Krankheit beiträgt. Die konservative Bearbeitung der Repeat-Sequenzen, welche die natürliche Intrinsizität und Lesbarkeit der Gene nicht beeinträchtigt, könnte das therapeutische Fenster für präventive und kurative Interventionen weit öffnen. Darüber hinaus werden ähnliche Prinzipien auch für andere neurodegenerative Trinukleotid-Repeat-Erkrankungen wie Friedreich-Ataxie diskutiert. Dort werden ebenfalls bewährte Basen-Editoren zur gezielten Modifikation der repetitiven DNA-Abschnitte eingesetzt, die für den Krankheitsverlauf maßgeblich sind.
Das Erfolgsmodell bei Huntington liefert somit einen zuverlässigen Prototyp für die breite Anwendung solcher molekularen Ansätze. Die Entdeckung, dass kleine Interruptionsmuster von CAA-Codons in der Trinukleotid-Region bei Patienten natürlich vorkommen und mit einer signifikant verzögerten Krankheitsmanifestation assoziiert sind, wurde durch Populationstudien und klinische Korrelationen umfassend unterstrichen. Diese genetische Variation bildet eine Art molekulare „Schutzschicht“ in der DNA, die die Instabilität und Ausdehnung der toxischen Repeat-Abschnitte vermindert. Durch Basen-Editing lässt sich ein solches Schutzmuster aktiv imitieren und therapeutisch realisieren. Wichtig ist, dass die derzeit verwendeten Methoden hauptsächlich auf Patientenmodellen in vitro und auf Jungtieren in vivo getestet wurden.
Es ergeben sich deshalb weitere Forschungsaufgaben, beispielsweise zur Langzeitwirkung der Interruptionsbearbeitung bei älteren Tieren, Macht und Grenzen der somatischen Repeat-Stabilisierung im Erwachsenenalter oder zur möglichen immunologischen Reaktion auf virale Vektorverabreichung. Auch gilt es, die biologische Bedeutung eventueller Off-Target-Variationen vertiefend zu klären, um die Sicherheit der Therapie vor klinischer Umsetzung uneingeschränkt gewährleisten zu können. Potenziale des Genom-Editings liegen zudem in der Verknüpfung mit neuen Vektor-Technologien, die verbesserte Gewebe-Spezifität oder kontrollierte Expression ermöglichen. Die Nutzung alternativer AAV-Serotypen oder RNA-basierten transienten Delivery-Systemen könnte zukünftig dazu beitragen, die Sicherheit und Effektivität der Repeat-Interruptionsbearbeitung gezielt zu optimieren. Insgesamt lässt sich festhalten, dass die präzise Unterbrechung von CAG-Trinukleotid-Repeats mittels Basen-Editing ein vielversprechender neuer Ansatz zur Verminderung der somatischen Expansionen bei Huntington ist.
Dies könnte den Krankheitsverlauf entscheidend verzögern oder verbessern. Die Kombination aus molekularer Präzision, gezielter Intervention am genetischen Ursprung und nachweislicher Wirksamkeit in Patientenzellen wie in Tiermodellen stellt einen Meilenstein in der Huntington-Forschung dar, der den Weg für neuartige therapeutische Strategien ebnet. Noch in der Zukunft liegt das Ziel, diese Ansätze weiter zu verfeinern und klinisch in Studien zu validieren. Erste Sicherheits- und Wirksamkeitsdaten werden mit Spannung erwartet. Der aktuelle Forschungsstand signalisiert, dass das Genom-Editing in seiner Präzision und Wirkstärke nicht nur den Grundstein für innovative Huntington-Therapien legt, sondern auch vielfältige Anwendungen bei weiteren Repeat-bedingten Erkrankungen ermöglichen kann.
Gerade für unheilbare neurodegenerative Leiden, wo derzeit nur symptomatische Behandlungen existieren, öffnet sich damit eine neue Ära personalisierter und ursachenorientierter Medizin.