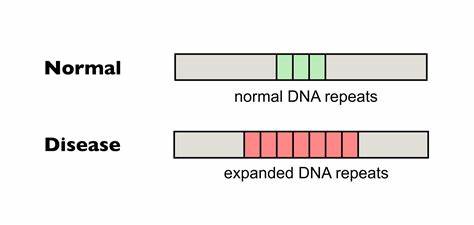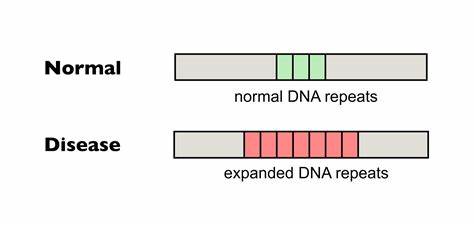
Huntington-Krankheit (HD) ist eine vererbte neurodegenerative Erkrankung, die durch die pathologische Expansion von CAG-Trinukleotid-Wiederholungen im HTT-Gen verursacht wird. Die Anzahl der CAG-Wiederholungen bestimmt maßgeblich den Krankheitsbeginn, den Verlauf und die Schwere der Symptome. Ein wesentlicher Mechanismus, der das Fortschreiten von HD beeinflusst, ist die somatische Instabilität – das heißt die weitere Ausdehnung der CAG-Trinukleotidwiederholungen in bestimmten Geweben, insbesondere im Gehirn, über die Lebenszeit eines Patienten hinweg. Die somatische Expansion verlängert die Polyglutamin-Strecke im Huntingtin-Protein, was die toxische Wirkung verstärkt und zur Neurodegeneration beiträgt. Bis vor Kurzem war dieses Phänomen eine unüberwindbare Hürde bei der Entwicklung wirksamer therapeutischer Ansätze.
Aktuelle Durchbrüche in der präzisen Genome-Editierung haben jedoch neue Wege eröffnet, die Wiederholungsausdehnung zu modulieren und potenziell den Krankheitsverlauf aufzuhalten oder abzuschwächen. Insbesondere innovative Basen-Editing-Technologien bieten die Möglichkeit, einzelne Nukleotide gezielt zu verändern, ohne dabei DNA-Doppelstrangbrüche hervorzurufen. Diese Methode ermöglicht es, an spezifischen Stellen innerhalb der CAG-Wiederholung ein häufig natürlich vorkommendes Synonym einzufügen – nämlich die CAA-Trinukleotidunterbrechung. Da CAG und CAA beide für Glutamin kodieren, bleibt das Protein unverändert, doch die genomische Stabilität der Sequenz wird deutlich erhöht. Zahlreiche Studien haben gezeigt, dass natürliche Unterbrechungen der CAG-Wiederholung, zum Beispiel durch CAA, mit einer verzögerten Krankheitsmanifestation und reduzierter somatischer Expansion einhergehen.
Die Einführung solcher Interruptions in langen CAG-Trakten wurde daher als vielversprechende therapeutische Strategie angesehen, um das Progressionsrisiko zu vermindern. Experimentelle Untersuchungen mit zellulären HD-Modellen konnten in vitro erfolgreich zeigen, dass Cytosin-Basen-Editoren (CBEs) mithilfe guidender RNA-Sequenzen an CAG-Trinukleotid-Wiederholungen binden und diese gezielt in CAA verändern können. In patientenabgeleiteten Fibroblasten mit pathologischer CAG-Expansion konnte damit eine signifikante Reduktion der somatischen Expansion beobachtet werden. Auf molekularer Ebene verhindern die CAA-Unterbrechungen die Bildung von stabilen DNA-Strukturierungen wie Haarnadelschleifen und R-Loops, die für die Fehleranfälligkeit während der DNA-Replikation und Transkription verantwortlich sind. Diese abnormalen Strukturen sind Ausgangspunkt für Reparaturprozesse, die zur Verlängerung der Wiederholungen beitragen.
Somit schützt die gezielte Interruptions-Editierung vor einer weiteren Instabilität des Genabschnitts. Tierstudien bestätigen die Übertragbarkeit dieser Ergebnisse in vivo. In humanisierten HD-Mausmodellen namens Htt.Q111, die eine pathogene CAG-Expansion im Huntingtin-Gen tragen, konnte die AAV9-vermittelte Übertragung von Basen-Editoren in den zentralen Nervensystem-Gehalt, insbesondere in Kortex und Striatum, erfolgversprechend eingeführt werden. Die in vivo durchgeführte CAG-zu-CAA-Editierung reduzierte nachweislich die somatische Expansion der pathogenen CAG-Wiederholung und führte zum Teil sogar zu einer Verkürzung der Wiederholungskette.
Dies lässt darauf schließen, dass eine frühzeitige gentechnische Intervention mit Unterbrechungen in der Wiederholung das Fortschreiten bzw. die Initiierung neurodegenerativer Prozesse hemmen kann. Neben der gezielten Effektivität ist auch die Sicherheitsbewertung ein zentraler Aspekt. Um potenzielle unerwünschte off-target Effekte zu evaluieren, wurden umfassende Genome-weit Analysen durchgeführt. Die Resultate zeigten, dass die Basen-Editoren bevorzugt auf den Zielsequenzen innerhalb der CAG-Wiederholungen wirken und Off-target-Modifikationen überwiegend in Regionen ohne bedeutsame Protein-codierende Funktionen auftreten.
Zudem resultieren off-target-Veränderungen meist in synonymen Mutationen, die weder der Aminosäuresequenz noch der Proteinfunktion nachteilig sind. In einzelnen Fällen können zwar nonsynonyme Veränderungen gefunden werden, deren Einfluss jedoch mit bioinformatischen Tools als überwiegend gering eingestuft wird. Dennoch bedarf es weiterführender Forschung, um mögliche Langzeitfolgen und Einfluss auf neuronale Funktionen sicher auszuschließen. Die Übertragung dieser Technologie auf andere TNR-Erkrankungen, wie beispielsweise die Friedreich-Ataxie, bei der ebenfalls eine somatische Expansion pathogener GAA-Repeats eine Rolle spielt, hat sich ebenfalls als effektiv herausgestellt. Adenin-Basen-Editoren (ABEs) ermöglichen die Einführung spezifischer Interruptions in die GAA-Sequenzen des FXN-Gens, wodurch auch dort somatische Expansionen reduziert und die Genexpression verbessert werden können.
Das Basen-Editing stellt somit eine innovative und zielgerichtete Methode dar, mit der sich potenziell die genetische Basis der Huntington-Erkrankung und verwandter TNR-assoziierter Krankheiten behandeln lässt. Die Erfolge in Zellkulturmodellen und animalischen Systemen legen nahe, dass durch frühzeitige und präzise Interventionen im Gehirn die Progression verlangsamt werden kann. Dies ist ein wesentlicher Schritt hin zu dauerhaften, krankheitsmodifizierenden Therapien, die über symptomatische Behandlungsansätze hinausgehen. Die klinische Umsetzung steht allerdings vor Herausforderungen. Die effiziente und sichere Lieferung der Basen-Editoren in das menschliche Gehirn, mögliche Immunreaktionen gegen virale Vektoren, sowie die langfristige Stabilität der Editierungen und Verträglichkeit müssen eingehend geprüft werden.
Auch die Erreichbarkeit weiterer Erkrankungsmodelle und Therapieindikationen sind Gegenstand intensiver Forschung. Zusammenfassend stellt die gezielte Unterbrechung von CAG-Repeat-Expansionen mittels präziser Basen-Editoren eine vielversprechende Strategie dar, um die somatische Instabilität in Huntington-Patientenzellen signifikant zu reduzieren. Die Verringerung der Repeat-Expansionen könnte den Krankheitsverlauf maßgeblich positiv beeinflussen, indem sie das Fortschreiten der Neurodegeneration verlangsamt oder sogar verhindert. Mit der weiteren technischen Optimierung und klinischen Erprobung könnte dieses Verfahren zukünftig ein wichtiger Bestandteil gezielter genetischer Therapien bei Huntington und verwandten TNR-Erkrankungen sein.