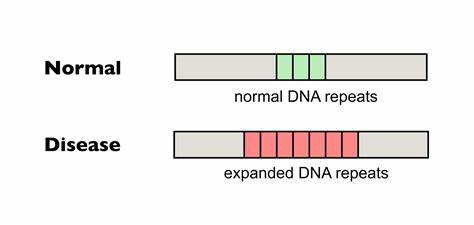
Die Huntington-Krankheit (HD) stellt eine der bekanntesten genetisch bedingten neurodegenerativen Erkrankungen dar und ist durch eine pathologische Expansion von CAG-Trinukleotid-Wiederholungen im Huntingtin-Gen charakterisiert. Diese langen Wiederholungen destabilisieren das Genom und führen zu einer schrittweisen Ausdehnung der Sequenz im Laufe des Lebens, was die Progression und den Schweregrad der Krankheit maßgeblich beeinflusst. Die somatische Expansion der CAG-Repeats stellt somit einen wichtigen Faktor in der Krankheitsentwicklung dar und wurde daher zum Fokus moderner therapeutischer Forschungsbemühungen. Eine innovative Strategie, die sich in den letzten Jahren zunehmend etabliert hat, ist die präzise Genom-Editierung mittels sogenannter Basen-Editoren. Diese Technologie ermöglicht es, einzelne Nukleotide gezielt umzuwandeln, ohne dabei das Erbgut vollständig zu durchtrennen oder größere Schäden zu verursachen.
Im Fall von HD konzentrieren sich die Forscher darauf, die reine Sequenz der CAG-Wiederholungen aufzubrechen und durch stabile Unterbrechungen wie die CAA-Codons zu ersetzen. Diese Unterbrechungen kommen natürlicherweise in der Bevölkerung vor und sind mit einer verminderten Instabilität der Repeat-Trakte sowie einem verzögerten Erkrankungsbeginn assoziiert. Dadurch bieten sie eine vielversprechende Möglichkeit, das Fortschreiten der Erkrankung zu verlangsamen oder gar zu verhindern. Aktuelle Studien haben gezeigt, dass der Einsatz von cytosine base editors (CBEs) erfolgreich ist, um gezielt CAG-Sequenzen im Huntingtin-Gen zu modifizieren. In humanen Fibroblasten von Patienten konnten bereits nach kurzer Zeit signifikante Einführungen von CAA-Unterbrechungen innerhalb der pathogenen Repeat-Trakte beobachtet werden.
Dabei zeigte sich, dass längere, krankheitsverursachende Repeatlängen besser und häufiger editiert wurden als kürzere, was auf eine verbesserte Bindungsmöglichkeit des Cas9-Komplexes an längere Repeat-Sequenzen zurückzuführen ist. Zudem konnte in Zellkulturversuchen nachgewiesen werden, dass diese gezielten Unterbrechungen die somatische Expansion der CAG-Wiederholungen effektiv hemmen. Während Kontrollzellen ohne Editierung im Zeitverlauf eine kontinuierliche Ausdehnung der Repeat-Länge zeigten, blieben die durch Basen-Editierung modifizierten Allele stabil oder wiesen sogar eine leichte Kontraktion auf. Dies unterstreicht die therapeutische Relevanz der Wiederholung, da eine Verhinderung der Expansion die Wahrscheinlichkeit einer frühen Krankheitsmanifestation deutlich senken kann. Neben zellulären Modellen wurde die Methode auch in vivo erfolgreich auf Mäuse übertragen, die ein humanisiertes Huntingtin-Gen mit pathologischer CAG-Wiederholung tragen.
Die Verwendung eines AAV9-basierten Vektorsystems ermöglichte die zielgerichtete Lieferung des Base-Editing-Systems in das zentrale Nervensystem der Neonatale. Die Ergebnisse dieser in-vivo-Anwendungen bestätigten die In-vitro-Beobachtungen: Es wurden signifikante Einfügungen von stabilisierenden CAA-Unterbrechungen in relevanten Hirnarealen wie Cortex und Striatum erreicht. Diese Änderung ging mit einer nachweislichen Reduktion der somatischen Repeat-Expansionen in den behandelten Geweben einher. Interessanterweise konnte die Expansion nicht nur stabilisiert, sondern teilweise sogar eine Verkürzung der Repeat-Länge festgestellt werden, was für eine aktive Umkehrung des pathologischen Prozesses spricht. Ein weiterer relevanter Aspekt dieser Therapie betrifft die Sicherheit und mögliche Off-Target-Effekte der Basen-Editoren.
Durch umfassende Genome-weite Analysen unter Verwendung von Methoden wie CIRCLE-seq und Hochdurchsatzsequenzierung konnte gezeigt werden, dass die meisten unerwünschten Editierungen in nicht kodierenden Bereichen des Genoms auftreten. Protein-kodierende Off-Target-Veränderungen wurden überwiegend als synonym charakterisiert, sodass sie den Aminosäure-Code nicht verändern und vermutlich keine negativen Auswirkungen auf Zellfunktionen haben. Nur ein geringer Anteil der Off-Target-Editierungen führte zu nicht-synonymen Mutationen, von denen wiederum die meisten von Predictive-Tools als vermutlich neutral bewertet wurden. Trotz dieser positiven Sicherheitsbewertung betonen die Forscher die Notwendigkeit weiterführender Studien, um die langfristigen Effekte detailliert zu untersuchen und mögliche Risiken auszuschließen. Die Bedeutung dieser Ergebnisse liegt nicht nur in der Behandlung der Huntington-Krankheit, sondern wirkt als Blaupause für den Umgang mit einer Vielzahl von Erkrankungen, die auf trinukleotidbedingter Geninstabilität beruhen.
Viele neurodegenerative Krankheiten, darunter verschiedene Spinocerebelläre Ataxien, teilen ähnliche Pathomechanismen, bei denen langsame somatische Repeat-Expansionen die Krankheit auslösen und verschlimmern. Das Konzept der gezielten Basen-Editierung, um natürliche, schützende Unterbrechungen in die schädlichen Repeats einzubauen, könnte daher potenziell auf zahlreiche weitere Erkrankungen übertragen werden. Zusätzlich zu den Aspekten der Basen-Editierung ist auch die Wahl der Lieferplattform entscheidend. AAV9 hat sich als effektives Vehikel erwiesen, um die zu therapierende DNA an das zentrale Nervensystem abzugeben. Dennoch bestehen Herausforderungen hinsichtlich der Einschränkungen bezüglich der Zielzellen, der Immunantwort und einer dauerhaften Expression des Editors.
Zukünftige Forschungsarbeiten widmen sich deshalb Entwicklung von Technologien, die eine gezieltere, möglicherweise auch temporäre Expression des Editors ermöglichen, um Nebenwirkungen weiter zu minimieren und die therapeutische Sicherheit zu verbessern. Insgesamt markieren diese Fortschritte einen bedeutenden Schritt in der Behandlung genetischer Erkrankungen und legen den Grundstein für zukünftige klinische Anwendungen. Die Fähigkeit, genomische Repeat-Expansionen direkt und präzise zu modifizieren, eröffnet neue Perspektiven, um tödlichen neurodegenerativen Erkrankungen wie Huntington eine effektivere Therapiemöglichkeit entgegenzusetzen. Mit der Weiterentwicklung von Genom-Editierungstechnologien und verbesserter Vektor-Optimierung könnte die Verhinderung somatischer Repeat-Expansionen zu einem realistischen Ziel werden, das den Krankheitsverlauf entscheidend beeinflusst oder sogar aufhält. Die Möglichkeit, Änderungen an den CAG-Sequenzen vorzunehmen, spricht auch das zentrale Problem bei der Huntington-Krankheit an: die Anzahl der Wiederholungen bestimmt vor allem die Krankheitsdauer und -schwere.
Das bedeutet, dass selbst eine teilweise Reduktion der Expansion ausreichen könnte, um das klinische Erscheinungsbild signifikant zu verzögern. Zudem weisen die Studien darauf hin, dass solche Editierungen nicht nur pathogene Progression stoppen, sondern auch eine Wiederherstellung der genetischen Stabilität fördern können, was neue therapeutische Optionen eröffnet. Zukünftige Arbeiten sollten sich verstärkt interdisziplinär mit den klinischen Auswirkungen beschäftigen. Die Translation der in-vitro- und präklinischen Modelle in menschliche Kliniken wird die Sicherheit, Effizienz und Nachhaltigkeit dieser Verfahren bestimmen. Daneben ist die Entwicklung möglichst zielgerichteter Editing-Systeme, die zelltypspezifisch arbeiten, ein zentrales Forschungsziel, ebenso wie die Kontrolle immunologischer Reaktionen auf virale Vektoren oder editingbezogene Proteine.
Die kumulierten Ergebnisse der Basen-Editing-Forschung bei Huntington unterstreichen die Rolle modernster molekularbiologischer Ansätze im Kampf gegen erblich bedingte neurodegenerative Erkrankungen. Was vor einigen Jahren noch als science-fictionhaft galt, ist heute eine greifbare Möglichkeit, die den Patienten eine Zukunft mit weniger Krankheitsschmerzen und längerer Lebensqualität versprechen kann.




![Current Continuation E2: Satnam Singh (Groq) [video]](/images/729A936D-DE31-45CB-BA44-0D2952927494)

