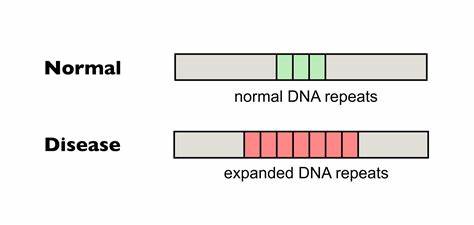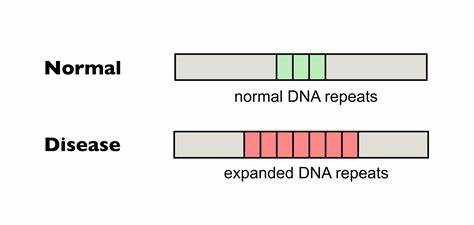
Die Huntington-Krankheit, eine genetisch bedingte neurodegenerative Störung, wird durch eine auffällige Erweiterung von DNA-Trinukleotid-Repeats im HTT-Gen verursacht. Diese Polyglutamin-Erkrankung führt unweigerlich zu schwerwiegenden motorischen, kognitiven und psychischen Beeinträchtigungen und betrifft weltweit tausende Menschen. Eine der zentralen molekularen Ursachen für die Pathogenese ist die somatische Instabilität der CAG-Repeat-Sequenzen, die dazu neigt, im Laufe des Lebens weiter zu expandieren. Diese somatischen Repeat-Expansionen verstärken die Neurodegeneration und beschleunigen den Krankheitsverlauf. Daher stellt die Begrenzung oder Verhinderung dieser Expansionen eine vielversprechende, therapeutische Zielsetzung dar.
Traditionelle Behandlungsansätze konzentrieren sich bislang auf symptomatische Linderung, während kausale Therapien bisher fehlgeschlagen sind. Die jüngsten Fortschritte in der Gentherapie und speziell in der Präzisionsgenomeditierung, wie der sogenannten Baseneditierung, eröffnen neue Horizonte. Im Gegensatz zu klassischen CRISPR-Cas9-Ansätzen, die Doppelstrangbrüche im DNA-Molekül erzeugen, ermöglichen Baseneditoren punktgenaue Einzelbasenumwandlungen ohne Bruch der DNA-Stränge. Dies reduziert Nebenwirkungen und minimiert unerwünschte genomische Veränderungen.In Bezug auf die Huntington-Krankheit wurde demonstriert, dass durch gezieltes Cytosin-zu-Thymin-Editing in den CAG-Regionen Stabilisierungen der Repeat-Strukturen erzielt werden können.
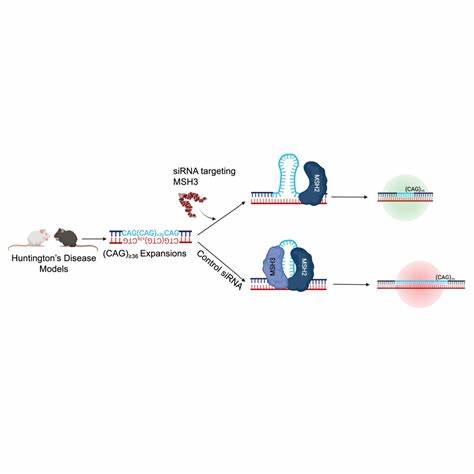
Durch die gezielte Einführung von sogenannten Unterbrechungen – also kleinen Nukleotidvariationen innerhalb der ansonsten reinen CAG-Repeatsequenz – erscheinen die Sequenzen weniger anfällig für weitere somatische Expansionen. Natürlich vorkommende Unterbrechungen, etwa CAA-Codons, die ebenfalls Glutamin codieren, werden mit milderen Krankheitsverläufen und verzögertem Ausbruch assoziiert. Die künstliche Einführung solcher Unterbrechungen durch Baseneditoren könnte somit die Genomstabilität verbessern und pathologische Expansionen eindämmen.Laborversuche an Patientenzellen illustrieren die Machbarkeit dieses Ansatzes. Hier wurden Fibroblasten von Huntington-Patienten mit Baseneditoren und spezifischen Leit-RNA-Sonden behandelt, die auf die CAG-Repeatbereiche zielgerichtet sind.
Die Folge waren signifikante Veränderungen im Repeat-Muster, mit einem bemerkenswerten Rückgang von somatischen Expansionen über Kultivierungszeitspannen von mehreren Wochen. Verbesserte Editierstrategien ermöglichen es, dass ein Großteil der Pathogenen Allele mehrere Unterbrechungen aufweist, was deren Instabilität nachweislich vermindert.Der Schritt von der Zellkultur zum lebenden Organismus wurde durch die Anwendung von Adeno-assoziierten Viren (AAV) als Vektoren für die Baseneditoren vollzogen. Neonatale Infektionen in gentechnisch veränderten Mausmodellen der Huntington-Krankheit resultierten in einer hohen Effizienz der Editierung in Zielgeweben wie dem Kortex und Striatum – Gehirnregionen, die bei der Huntington-Krankheit besonders betroffen sind. Die editierte Population zeigte deutlich reduzierte somatische Repeat-Expansionen, sowohl qualitativ als auch quantitativ hochsignifikant.
Diese Ergebnisse unterstreichen die biologisch relevante Auswirkung der neuen Therapieprinzipien.Eine besondere Herausforderung liegt im Umgang mit möglichen Off-Target-Effekten, also ungewollten Mutationen an anderen Stellen im Genom. Genome-weit durchgeführte Untersuchungen zum Off-Target-Editing belegten überwiegend geringe und meist unschädliche Veränderungen, häufig in nichtkodierenden Bereichen oder als sogenannte synonyme Mutationen, die keine Auswirkungen auf das Protein haben. Die sorgfältige Validierung und Optimierung der Baseneditoren sowie der Leit-RNAs ist deshalb von essenzieller Bedeutung, um die Sicherheit zukünftiger Therapien zu gewährleisten.Neben der Anwendung bei der Huntington-Krankheit zeigen ähnliche Ansätze, wie zum Beispiel die Adeninbaseneditierung bei Friedreich-Ataxie, dass die Reduktion somatischer Repeat-Expansionen durch Unterbrechungen der trinukleotidischen Sequenzen eine breite therapeutische Relevanz besitzt.
Die Reduktion der somatischen Instabilität trägt in beiden Fällen zu einer Stabilisierung der genetischen Grundlage bei und lässt hoffen, dass langfristig neurologische Schäden vermindert werden können.Insgesamt markieren diese Entwicklungen im Gen-Editing eine bedeutende Wende im Kampf gegen Huntington und verwandte Erkrankungen. Statt nur Symptome zu behandeln, wird nun direkt an der molekularen Ursache angesetzt, mit dem Ziel, die krankheitsauslösenden DNA-Mutationen zu modifizieren und zu stabilisieren. Für Patientinnen und Patienten könnte dies bedeuten, dass zukünftige Therapien das Fortschreiten der Krankheit verlangsamen oder gar verhindern und somit Lebensqualität und Lebenserwartung deutlich verbessern.Die nächsten Schritte umfassen klinische Studien zur Anwendung bei Menschen, die Optimierung der Vektoren für eine möglichst zielgenaue und effiziente Gentherapie sowie Langzeitbeobachtungen zur Sicherheit und Wirksamkeit.
Weiterhin gilt es, die Möglichkeiten alternativer Lieferwege für Baseneditoren zu erforschen, um möglichst viele betroffene Gewebe zu erreichen.Diese vielversprechende Strategie im Bereich des Gen-Editings bringt erneut Hoffnung in die Huntington-Forschung und setzt einen wichtigen Impuls in der Entwicklung personalisierter, genetisch-basierter Therapien gegen neurodegenerative Erkrankungen.