Die Huntington-Krankheit (HD) stellt eine der schwerwiegendsten neurodegenerativen Erkrankungen dar, die durch eine pathologische Expansion von CAG-Wiederholungen im Huntingtin-Gen (HTT) verursacht wird. Dieses Wiederholungssequenzmuster führt zu einer toxischen Verlängerung von Polyglutamin-Strecken im Huntingtin-Protein, was letztlich zu neuronaler Schädigung und klinischen Symptomen wie Bewegungsstörungen, kognitiven Beeinträchtigungen und Persönlichkeitsveränderungen führt. Trotz intensiver Forschung gibt es bislang keine kurative Behandlung, die den zugrunde liegenden Prozess der CAG-Expansion in den somatischen Zellen wirksam hemmen kann. Doch jüngste Fortschritte im Bereich der präzisen Genom-Editierung eröffnen neue therapeutische Perspektiven. Im Zentrum dieser Innovationen steht die Technologie der Baseneditierung, die es erlaubt, einzelne Basen im DNA-Strang gezielt und ohne Doppelstrangbrüche zu verändern.
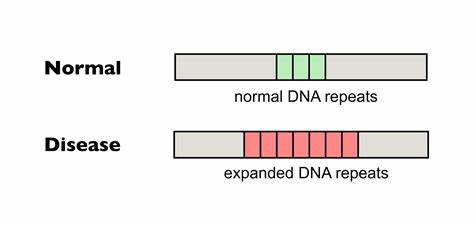
Forscher haben diese Technologie genutzt, um gezielt in die CAG-Trinukleotid-Regionen einzuwirken und sogenannte Interruptions—unterbrechende Nukleotidvarianten—zu integrieren, die natürlicherweise bei einigen Menschen vorkommen und mit einer stabileren Repeat-Struktur assoziiert sind. Insbesondere die Einführung von CAA-Unterbrechungen im CAG-Trakt kann die Instabilität der Repeats verringern und so die somatische Expansion, die im Verlauf der Huntington-Krankheit eine zentrale Rolle spielt, reduzieren. Die somatische Expansion von CAG-Repeats in Gehirnzellen, insbesondere in den Neuronen des Striatums und der Großhirnrinde, gilt als ein maßgeblicher Faktor für den Krankheitsausbruch und die Progression. Untersuchungen haben gezeigt, dass mit zunehmendem Alter die Zahl der CAG-Wiederholungen in einzelnen Zellen steigen kann, was zur Verschlimmerung der Krankheit beiträgt. Diese Instabilität ist jedoch nicht unausweichlich, denn natürliche Interruptionsvarianten, die einzelne CAG-Codons durch CAA ersetzen, sind mit einer reduzierten somatischen Expansion und einer verzögerten Krankheitsmanifestation verbunden.
Basierend auf diesem Konzept haben Wissenschaftler Cytosin-Baseneditoren (CBEs) entwickelt, die in der Lage sind, gezielt C in T (bzw. G in A auf der komplementären DNA-Strang) umzuwandeln und so CAG-Sequenzen in stabilere CAA-Sequenzen zu verwandeln, ohne die Aminosäuresequenz des Huntingtin-Proteins zu verändern. In vitro Experimente in Patientenfibroblasten zeigten, dass nach Anwendung des CBE-Systems ein signifikanter Anteil der HTT-Allele durch diese Interruptionsmutationen stabilisiert wurde. Besonders bemerkenswert war, dass nach mehreren Zellteilungsgenerationen in den behandelten Zellen eine deutlich verringerte somatische Expansion der CAG-Wiederholungen nachweisbar war, im Gegensatz zu unbehandelten Kontrollproben, die eine fortschreitende Verlängerung zeigten. Darüber hinaus wurden diese Baseneditoren erfolgreich in vivo in einem Htt.
Q111-Mausmodell angewandt, das eine humane Huntingtin-Variante mit pathologisch verlängerter CAG-Repeatsequenz exprimiert. Die virale Lieferung von optimierten CBEs mittels adenoassoziierter Viren (AAV9), gezielt ins Zentralnervensystem injiziert, führte bei neugeborenen Mäusen zur dauerhaften Installation von CAA-Interruptionsmutationen. Diese Intervention resultierte in einer signifikanten und nachhaltigen Reduktion der somatischen CAG-Expansion in Hirnregionen, die für die Huntington-Pathologie besonders relevant sind. Die Ergebnisse zeigten zudem, dass diese Bearbeitung nicht nur die Expansion verhinderte, sondern teilweise auch eine Verkürzung der Repeats bewirken konnte, was für zukünftige therapeutische Strategien von hoher Bedeutung ist. Ein wesentlicher Aspekt bei der Entwicklung dieser Ansätze ist die Analyse möglicher Off-Target-Effekte.
Zwar besteht theoretisch ein Risiko, dass Baseneditoren auch an nicht gewünschten Stellen im Genom tätig werden und dort ungewollte Mutationen induzieren, doch umfassende Studien mittels Ganzgenomsequenzierung und hochauflösenden Off-Target-Analysen zeigten, dass der Großteil der erzeugten Mutationen entweder in nicht-kodierenden Bereichen liegt oder durch synonyme Substitutionen keinen Einfluss auf die Proteinfunktion hat. Dennoch sind weiterführende Untersuchungen nötig, um die langfristige Sicherheit einer solchen Intervention zu beurteilen. Neben der Huntington-Krankheit wurden vergleichbare Strategien auch für andere trinukleotidbasierte Erkrankungen wie die Friedreich-Ataxie erforscht. Hier kommt ein Adenin-Baseneditor (ABE) zum Einsatz, der A-T Basenpaarungen gezielt in G-C Paare umwandelt und somit Interruptionsvarianten in langen GAA-Trinukleotidsträngen generiert. Auch in Patienten-Zellen und Mausmodellen konnte diese Methode die somatische Expansion der Wiederholungen vermindern und eine Erhöhung der Frataxin-Expression bewirken, was für das Krankheitsbild der Friedreich-Ataxie entscheidend ist.
Die therapeutische Relevanz dieser Forschung zeigt sich vor allem darin, dass die Reduktion der somatischen Repeat-Expansion potenziell eine Verzögerung des Krankheitsbeginns und eine Verlangsamung der Progression bewirken kann. Epidemiologische und molekulargenetische Daten unterstützen dieses Konzept, da indigene Interruptionsvarianten in Wiederholungssequenzen beim Menschen mit milderen Symptomen und späterem Ausbruch von Huntington und verwandten Erkrankungen assoziiert sind. Für die klinische Umsetzung der Baseneditor-basierten Therapie sind allerdings noch einige Herausforderungen zu bewältigen. Dazu gehört neben der weiteren Optimierung der Editoreffizienz und Spezifität vor allem die Entwicklung geeigneter Verabreichungswege. Die im Mausmodell verwendete AAV9-basierte intrazerebroventrikuläre Injektion ist derzeit nicht ohne Weiteres auf den Menschen übertragbar.
Alternative Ansätze wie die Entwicklung von weniger immunogenen Vektorplattformen, viralen Varianten mit erhöhter Gewebespezifität, oder nicht-virale Partikelsysteme könnten zukünftig dazu beitragen, eine sichere, langfristige und effektive Gentherapie zu ermöglichen. Ein weiteres wichtiges Thema ist die Langzeitwirkung der DNA-Base-Editing-Therapien. Da AAV-Vektoren eine anhaltende Expression der Baseneditorproteine bewirken, ist die Kontrolle über die Dauer und Ausprägung des Editings im Patientengenom essenziell, um unerwünschte Nebeneffekte zu minimieren. Forschungsansätze beschäftigen sich daher momentan mit transienten Expressionstechnologien und gezieltem Zieltissue-Spezifität. Die bisherigen Ergebnisse der somatischen Interruptions-Editierung demonstrieren darüber hinaus einen vielversprechenden Proof-of-Concept für eine sehr neue Klasse von Gentherapien bei neurodegenerativen Repeat-Erkrankungen.
Die Verlagerung des therapeutischen Ansatzes vom Symptombehandeln hin zur genetischen Prävention oder Verlangsamung der somatischen Expansion stellt eine paradigmatische Veränderung dar. Zusammenfassend zeigt die gezielte Einführung von Unterbrechungen in pathogene Trinukleotid-Repeatregionen durch Cytosin- oder Adenin-Baseneditoren ein enormes Potenzial zur Stabilisierung dieser genetischen Elemente. Durch die Verringerung der somatischen Expansion in relevanten Zelltypen kann der progressive Verlust der neuronalen Funktionen, der bei Huntington und verwandten Erkrankungen beobachtet wird, vermutlich aufgehalten oder zumindest verlangsamt werden. Die derzeitigen Studien liefern eine fundierte Basis für die Weiterentwicklung dieser Technologie und für zukünftige klinische Anwendungen, die das Leben vieler Patienten nachhaltig verbessern könnten.