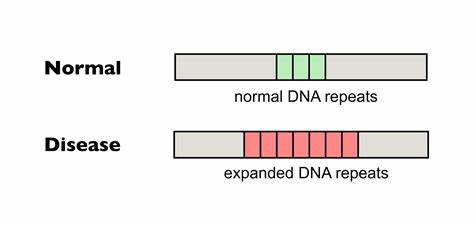
Die Huntington-Krankheit (HD) gehört zu den sogenannten trinukleotid-Repeat-Erkrankungen, welche durch die pathologische Ausweitung von dreierbasigen DNA-Sequenzen in bestimmten Genen gekennzeichnet sind. Bei HD speziell handelt es sich um die Expansion von CAG-Wiederholungen im Huntingtin-Gen (HTT), die zu einer toxischen Verlängerung der Polyglutamin-Botenstoffkette im Huntingtin-Protein führt. Die Anzahl der CAG-Wiederholungen bestimmt dabei wesentlich den Schweregrad und das Erkrankungsalter. Je länger die Wiederholungssequenz, desto früher und schwerer manifestiert sich die Krankheit. Ein besonderes Merkmal dieser Störung ist jedoch die somatische Instabilität der Repeatregionen, bei welcher sich die Anzahl der Wiederholungen im Laufe des Lebens in verschiedenen Körperzellen weiter verändern kann.
Dies führt zu zunehmender DNA-Schädigung und verstärkt den neurodegenerativen Prozess. Die somatische Expansion der CAG-Wiederholungen in betroffenen Patienten ist ein kritischer Faktor für die Progression der Erkrankung. Forschungen haben gezeigt, dass vor allem Nervenzellen in Schlüsselbereichen des Gehirns wie dem Striatum und dem Cortex diese Erweiterungen im Verlauf des Lebens ansammeln. Somit gelangt ein Großteil der Zellen über einen längeren Zeitraum hinweg zunehmend in einen krankheitsfördernden Zustand, der am Ende neuronale Degeneration und den Verlust motorischer sowie kognitiver Fähigkeiten nach sich zieht. Bislang gab es keine zugelassene Therapie, die gezielt auf diese genetische Ursache und insbesondere auf die somatische Repeat-Erweiterung abzielt.
Konventionelle Ansätze fokussieren sich meist auf symptomatische Behandlung oder die Reduktion der Huntingtin-Proteinproduktion. Die jüngsten Fortschritte auf dem Gebiet der Gentechnik und insbesondere der Basenbearbeitung eröffnen jedoch eine neue vielversprechende Möglichkeit, direkt in die DNA-Sequenz der repetitiven Abschnitte einzugreifen. Die Basenbearbeitung ist eine innovative molekulare Methode, bei der einzelne Nukleotidbasen in der DNA gezielt und präzise verändert werden können, ohne dass dabei Doppelstrangbrüche im Erbgut entstehen. Bei der Huntington-Krankheit bietet sich insbesondere die Einführung von sogenannten Interruptionssequenzen in den CAG-Repeattrakt an. Durch den gezielten Austausch einzelner Nukleotide können ununterbrochene CAG-Wiederholungen in ein Gemisch aus CAG und CAA umgewandelt werden, wobei beide Codons für Glutamin kodieren.
Diese CAA-Unterbrechungen sind in manchen natürlichen, nicht-pathogenen Varianten vorhanden und zeichnen sich dadurch aus, dass sie die Stabilität der Repeatregion erhöhen und die somatische Expansion deutlich verringern. Forschungen mit patientenabgeleiteten Fibroblasten sowie in tierischen Modellen haben gezeigt, dass die Anwendung von Cytosin-Baseneditieren (CBEs) zur Einführung von CAA-Unterbrechungen die somatische Repeat-Expansion signifikant reduziert. In vitro konnten dadurch nach nur wenigen Tagen bis Wochen eine hohe Effizienz an Interruptionsinstallationen erreicht werden, die mit verminderter Instabilität der Repeatregion einhergingen. Auch in vivo führte die Lieferung solcher Baseneditoren mittels AAV9-Vektoren bei Mäusen mit humanisierten HTT-Genen zu einer effizienten Bearbeitung der Zielregion in verschiedenen Hirnarealen und einer signifikanten Verringerung der somatischen Verlängerung im Vergleich zu unbehandelten Kontrolltieren. Die Vorteile dieser neuen Methode liegen vor allem in der dauerhaften Stabilisierung der Repeatregion und der damit einhergehenden möglichen Verzögerung oder sogar Verhinderung des Auftretens von Symptomen sowie der Verlangsamung des Krankheitsverlaufs.
Zudem können durch die präzise Bearbeitung potenzielle unerwünschte Effekte, wie etwa Indel-Mutationen oder unerwünschte Protein-Veränderungen, minimiert werden. Um eine sichere Anwendung zu gewährleisten, wurde in umfangreichen Off-Target-Analysen festgestellt, dass die meisten unerwünschten Editierungen in nicht-codierenden oder synonymen Bereichen auftreten und somit keine nachteiligen Auswirkungen auf die Proteinfunktion haben. Ebenso sprechen Daten aus Zellkulturen und Tiermodellen dafür, dass länger bestehende Interruptionsmotive den Expansionsprozess schon bei reduzierten Mengen an Baseneditoren hemmen können. Dieses Phänomen ergibt sich daraus, dass ununterbrochene repetitive Sequenzen besonders anfällig für fehlgeleitete DNA-Reparaturprozesse sind, welche die Expansion auslösen. Die Integration von Unterbrechungen stabilisiert diese Regionen, was die Bildung komplexer DNA-Strukturen vermindert und die Kontrolle durch zelluläre Reparatursysteme erleichtert.
Im Hinblick auf zukünftige klinische Anwendungen müssen hierzu jedoch noch mehrere Herausforderungen bearbeitet werden. Neben der effizienten, gezielten und möglichst schonenden Lieferung der Baseneditoren in die relevanten menschlichen Hirnregionen stellt die Sicherheit der Technologie weiterhin einen zentralen Aspekt dar. Die Entwicklung besserer und weniger immunogenen Träger-Systeme sowie die Etablierung präziser Dosierungs- und Kontrollmethoden sind für die Translation in die Klinik entscheidend. Zusätzlich ist die tiefere Untersuchung von Langzeiteffekten nach Editierung notwendig, um sowohl die fortdauernde Wirksamkeit als auch potenzielle Nebenwirkungen zu bewerten. Die Erkenntnisse zu Interruptionssequenzen bieten zudem Einblicke in die molekularen Mechanismen von HD und anderen trinukleotid-bedingten Erkrankungen.
Sie zeigen, wie besondere DNA-Varianten das Risiko und die Ausprägung der Erkrankung modulieren können. Daraus ergibt sich die Perspektive, dass therapeutische Strategien, welche repeat-Untereichnungen genetisch nachahmen, das Fortschreiten verschiedener TNR-Erkrankungen grundlegend verändern und verbessern könnten. Parallel zu den Arbeiten am HTT-Gen wird auch die gezielte Basenbearbeitung bei anderen genetischen Erkrankungen mit trinukleotidischen Expansionen untersucht, wie beispielsweise der Friedreich-Ataxie. Dabei wird deutlich, dass die Prinzipien der Interruptionsinduzierten Stabilisierung universell anwendbar sind, was einen Paradigmenwechsel für die Behandlung dieser genetisch bedingten neurodegenerativen Erkrankungen bedeuten könnte. Zusammenfassend eröffnet die gezielte Basenbearbeitung der CAG-Wiederholungen im HTT-Gen eine aussichtsreiche neue therapeutische Richtung bei der Huntington-Krankheit.
Die Fähigkeit, somatische Expansionen in patienteneigenen Zellen und in vivo signifikant zu reduzieren, weist auf das Potenzial hin, den neurodegenerativen Verlauf zu bremsen oder teilweise zu verhindern. Während noch umfangreiche weitere Forschung für eine klinische Anwendung notwendig ist, markieren diese Fortschritte einen wichtigen Meilenstein hin zu personalisierten und molekular zielgerichteten Behandlungen von Huntington und verwandten Repeat-Erkrankungen.