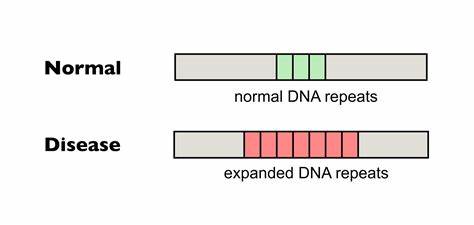
Huntington-Krankheit (HD) gehört zu den sogenannten trinukleotid-Repeat-Erkrankungen, bei denen eine abnormale Ausweitung kurzer Wiederholungssequenzen im Erbgut die Ursache für die fortschreitende neurodegenerative Symptomatik ist. Besonders die verlängerten CAG-Repeats im Huntington-Gen (HTT), welche die Bildung toxischer Polyglutamin-Ketten bewirken, sind hierbei von zentraler Bedeutung. Der Grad der Erkrankungskrankheit, das Alter beim Ausbruch und die Geschwindigkeit der Progression korrelieren eng mit der Anzahl der CAG-Repeat-Einheiten, die sich jedoch im Verlauf des Lebens durch somatische Instabilität weiter vergrößern können. Diese somatische Expansion von Wiederholungen in neuronalen und anderen Geweben verschärft den Krankheitsverlauf und stellt somit ein attraktives therapeutisches Ziel dar. In den letzten Jahren haben innovative Methoden der Genom-Editierung, insbesondere die Entwicklung sogenannter Base-Editoren, neue Wege aufgezeigt, um gezielt und präzise einzelne Basenpaare im Erbgut zu verändern, ohne die DNA doppelt zu schneiden.
Durch diese Technologien kann man Veränderungen vornehmen, die beispielsweise natürliche Unterbrechungen innerhalb von stark repetitiven Sequenzen nachahmen, welche das genetische Material stabilisieren und somatische Expansionen hemmen können. Ein kürzlich erschienener Meilenstein in der Forschung demonstriert, wie die gezielte Einfügung von CAA-Unterbrechungen in die pathogenen CAG-Repeat-Trakte bei Huntington mittels cytosiner Baseneditoren (CBEs) die somatische Expansion in patienteneigenen Zellen sowie in Tiermodellen signifikant reduziert. Diese Unterbrechungen sind natürlich vorkommende Varianten, die bei einzelnen Individuen mit verlängerter Lebenserwartung und verzögertem Krankheitsbeginn assoziiert sind. Die Studie weist überzeugend nach, dass die Anwendung eines präzisen Cytosin-Baseneditors, kombiniert mit einem auf CTG-Repeatsequenzen angepassten guide RNA (sgRNA), in patienteneigenen Fibroblasten eine hohe Effizienz bei der Erzeugung gezielter CAG-zu-CAA-Konversionen aufweist. Dieses editierende Verfahren führte nicht nur zur Stabilisierung der Repeatlänge sondern zeigte über einen Zeitraum von mehreren Wochen eine deutliche Verhinderung der sonst typischen somatischen Expansion.
Die Besonderheit liegt darin, dass diese Editierungen synonym sind und keine Aminosäuresequenzänderungen verursachen, was unerwünschte Nebeneffekte verringert. Darüber hinaus konnten diese Ergebnisse in vivo durch die virale Auslieferung eines optimierten CBEs mittels AAV9-Vektor in neonatalen Htt.Q111-Mäusen bestätigt werden. Dort wurden deutliche Reduktionen der CAG-Repeatlänge in zentralnervösen Geweben beobachtet, die für die HD-Pathologie relevant sind. Die Effizienz der Baseneditierung nahm im Verlauf von Wochen sogar weiter zu, was auf die anhaltende Expression der Base-Editor-Komponenten durch den Vektor zurückzuführen ist.
Ein wichtiges Sicherheitsmerkmal der verwendeten Base-Editing-Strategie ist der niedrige Off-Target-Effekt. Genomeweite Analysen mittels CIRCLE-seq und hochauflösender Ganzgenomsequenzierung zeigten, dass die meisten unerwünschten Modifikationen in nicht-codierenden Regionen oder in Synonymen Mutationen auftreten, die die Proteinsequenz nicht beeinflussen. Nur ein kleiner Teil der Off-Target-Bearbeitungen führte zu potenziell relevanten Proteinveränderungen, was jedoch weitere Untersuchungen zur klinischen Sicherheit erforderlich macht. Das Prinzip, pathogene Repeatsequenzen gezielt durch Einsprengungen natürlicher Unterbrechungen zu stabilisieren, lässt sich auf weitere Repeat-Erkrankungen übertragen. Interessanterweise demonstrierte das Team auch eine Adenin-Baseneditierung (ABE) zur Einführung von GGA/GAG-Unterbrechungen in die GAA-Repeats des FXN-Gens, das bei Friedreich-Ataxie (FRDA) eine zentrale Rolle spielt.
Auch hier zeigte sich eine signifikante Reduktion der somatischen Expansionen in entsprechenden Patientenzellen und Mausmodellen. Diese Fortschritte bieten einen vielversprechenden Einblick in die präzise, genetisch basierte Therapie von TNR-Erkrankungen, welche bisher therapeutisch nur symptomatisch behandelbar waren. Die Fähigkeit, die molekulare Ursache, also die Instabilität des Repeattrakts im Genom, direkt zu adressieren, stellt einen Paradigmenwechsel dar. Herausforderungen bestehen dennoch hinsichtlich der gezielten und effizienten Auslieferung der Base-Editoren in menschliche Gewebe insbesondere im zentralen Nervensystem, der Langzeitwirkung und möglichen immunologischen Reaktionen auf virale Vektoren. Die AAV9-Vektoren besitzen zwar eine neuronenspezifische Tropismus, adressieren jedoch nicht alle von der Huntington-Pathologie betroffenen Zelltypen beziehungsweise andere Gewebe.
Zudem ist das Risiko für unerwünschte Off-Target-Editierungen sorgfältig weiter zu evaluieren. Ein weiterer Aspekt ist die Wirkung der Unterbrechungen auf die Krankheitsprogression bei bereits manifesten HD-Patienten. Ob die Verhinderung oder Umkehrung der somatischen Expansion die klinische Symptomatik signifikant verzögern oder gar stoppen kann, wird in zukünftigen Studien zu klären sein. Erste Daten lassen jedoch darauf schließen, dass somatische Expansionen über Jahre stattfinden bevor neuronale Degeneration einsetzt, was ein günstiges therapeutisches Zeitfenster bietet. Aus wissenschaftlicher Perspektive zeigen diese Ergebnisse auch neue Einblicke in die molekulare Pathogenese von Huntington.
Die stabilisierende Wirkung von Unterbrechungen durch einzelne Nukleotidsubstitutionen unterstützt die Hypothese, dass die Entstehung höherer DNA-Strukturformationen innerhalb homogener Repeatsektoren Auslöser der Repeatinstabilität ist. In der Folge sind die DNA-Reparaturmechanismen an somatischen Expansionen beteiligt und bieten weitere Interventionsansätze. Zusammenfassend lässt sich sagen, dass die präzise Base-Editing-Technologie einen vielversprechenden Ansatz zur Behandlung der Huntington-Krankheit und ähnlicher trinukleotid-Repeat-Erkrankungen darstellt. Die Einführung natürlicher, stabilisierender Repeat-Unterbrechungen in das pathogene Allel reduziert nachweislich die somatische Expansion in menschlichen Zellen und Tiermodellen. Dies stellt eine potenzielle therapeutische Strategie dar, die sowohl symptomatische Verschlechterungen aufhalten als auch womöglich die zugrundeliegende genetische Ursache mindern kann.
Die nächsten Schritte werden die Optimierung der Editierungseffizienz, Minimierung von Off-Target-Risiken sowie umfassende Prüfung der Langzeitsicherheit und Wirksamkeit in relevanten Patientenmodellen sein. Fortschritte in den Bereichen Virusvektor-Engineering, nicht-virale Liefermethoden und basierte Kodierungsoptimierung werden den Weg für mögliche klinische Anwendungen weiter ebnen. In der Summe markieren die Forschungsergebnisse einen bedeutenden Wendepunkt in der Huntington-Therapie und eröffnen neue Perspektiven für Patienten, die bisher wenig Hoffnung auf Behandlung hatten. Die Technologie der Baseneditierung könnte zukünftig den Umgang mit einer Vielzahl genetisch bedingter neurodegenerativer Erkrankungen revolutionieren und wissenschaftliche sowie medizinische Meilensteine setzen.