Die Erforschung der menschlichen Genomstruktur hat in den letzten Jahrzehnten bemerkenswerte Fortschritte gemacht, welche unser Verständnis der genetischen Regulation und der Krankheitsentstehung vertiefen. Besonders relevant ist dabei die Betrachtung des Genoms nicht nur als lineare Abfolge von DNA, sondern als dreidimensional organisiertes Molekül, das innerhalb des Zellkerns komplex gefaltet und arrangiert ist. Diese räumliche Organisation ermöglicht einen gezielten Austausch zwischen entfernten genomischen Regionen, die so als funktionelle Einheiten miteinander kommunizieren können. Im Kontext von Krebs hat sich gezeigt, dass diese dreidimensionale Genomlandschaft eine wesentliche Rolle spielt, indem sie die Aktivierung oder Hemmung von Onkogenen und Tumorsuppressorgenen steuert und somit entscheidend zur Tumorentwicklung sowie zum Fortschreiten der Krankheit beiträgt. Die dreidimensionale Genomorganisation umfasst verschiedene architektonische Ebenen wie A- und B-Kompartimente, die breite Muster der Chromatinumgebung repräsentieren, sowie Topologically Associating Domains (TADs), die niedrigere Strukturen bilden und Wechselwirkungen innerhalb begrenzter Genomabschnitte ermöglichen.
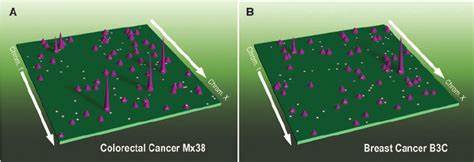
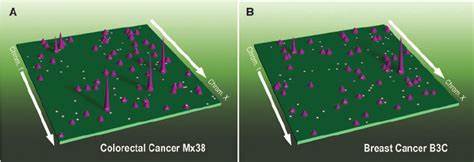
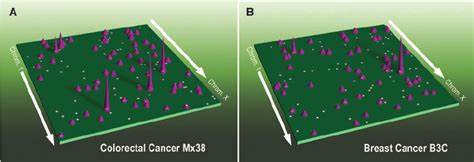
Besonders interessant sind hierbei die sogenannten Enhancer-Promoter-Interaktionen, die distal gelegene Regulatoren mit den Genpromotoren verbinden und so eine präzise Steuerung der Genexpression ermöglichen. In normalen Zellen sind diese Interaktionen hochspezifisch auf Zelltypen und den jeweiligen physiologischen Zustand abgestimmt. In Krebszellen jedoch kommt es häufig zu einer Umstrukturierung dieser Netzwerke, was als Enhancer-Rewiring bezeichnet wird und zu einer fehlerhaften Genregulation führt. Eine aktuelle umfassende Studie analysierte mithilfe der HiChIP-Technologie, die zugleich Proteinbindung und Chromatinstruktur misst, 69 Tumorgewebeproben aus 15 verschiedenen primären Krebsarten. Dabei wurde der Fokus auf das Histonmark H3K27ac gelegt, das aktive Enhancer und Promotoren kennzeichnet.
Das Ergebnis war eine beeindruckende Karte von über 665.000 signifikanten chromosomalen Interaktionen, welche die Vielfalt und Komplexität der dreidimensionalen Genomlandschaft in Krebszellen offenlegte. Die Daten zeigten, dass die Grundarchitektur der Kompartimente und TADs überwiegend erhalten bleibt, jedoch die feinere Ebene der Enhancer-Promoter-Loops stark variabel und abhängig vom Krebsgewebe ist. Diese Variabilität hat direkte funktionelle Konsequenzen für die Aktivität von Onkogenen. So wurden drei archetypische Muster der Enhancer-Nutzung identifiziert: Erstens stabile, d.
h. unveränderte Enhancer-Promoter-Beziehungen, zweitens selektive Gewinn von neuen Enhancer-Kontakten in bestimmten Krebsarten und drittens dynamische Umstrukturierungen, bei denen die Enhancer-Verkabelung zwischen den Genen stark variiert. Insbesondere für über 100 bekannte Onkogene wurde dieser Zusammenhang detailliert analysiert, was zeigte, dass bestimmte onkogene Aktivierungen nahezu ausschließlich durch eine Veränderung der dreidimensionalen Genomkonformation erklärt werden können. Darüber hinaus hat die Studie die Rolle genomischer struktureller Variationen (SVs) in der Krebsgenomlandschaft ins Visier genommen. Diese SVs, zu denen Deletionen, Duplikationen, Inversionen und Translokationen zählen, verändern nicht nur die lineare DNA-Ordnung, sondern haben auch drastische Auswirkungen auf die räumliche Genomorganisation.
Durch die Verschiebung von Enhancern in die Nähe neuer Zielgene fördern sie eine sogenannte Enhancer-Hijacking, wodurch sonst inaktive oder geringfügig aktive Genregulatoren plötzlich starke Tumorförderer aktivieren können. Bemerkenswert ist, dass extrachromosomale DNA (ecDNA), eine Art kreisförmige DNA-Struktur, die in einigen Krebsarten häufig vorkommt, eine besonders ausgeprägte Umgestaltung der Enhancer-Konnektivität bewirkt und somit zur massiven Überexpression von Onkogenen beiträgt. Neben den strukturellen Veränderungen spielen auch nichtkodierende Punktmutationen in Enhancer-Regionen eine wichtige Rolle in der Krebsgenomregulation. Die Analyse der Allelfrequenzen von Mutationen innerhalb aktiver Enhancer im Vergleich zum gesamten Genom zeigte, dass bestimmte somatische Mutationen die Aktivierung von Enhancern bewirken. Diese Mutationen können durch die Schaffung neuer Transkriptionsfaktorbindungsstellen oder die Verstärkung bestehender Motive eine erhöhte Genexpression auslösen.
Beispiele hierfür sind Mutationen im MECOM-Promotor bei Magenkrebs oder in Enhancern des FGFR1-Gens bei Blasenkarzinomen, die mit gesteigerter Onkogenexpression und schlechterer Prognose assoziiert sind. Ein weiterer bedeutender Aspekt ist die Deconvolution der dreidimensionalen Genomlandschaft von Krebsgewebe, das aus einer Heterogenität verschiedener Zelltypen besteht – Tumorzellen, Immunzellen, Stroma und andere Bestandteile des Tumormikromilieus. Mittels Integration von HiChIP-Daten mit Einzelzell-ATAC-seq-Daten konnte die Studie differenzieren, welche Enhancer-Promoter-Interaktionen in malignen Zellen und welche in immunologischen Zellen vorherrschen. Diese Analyse führte beispielsweise zur Entdeckung eines myeloidspezifischen Enhancers, der regulatorische Kontrolle über das Immune-Checkpoint-Gen CD274 (PD-L1) besitzt, und unterstreicht somit die Bedeutung von Zelltyp-spezifischen Genregulationsnetzwerken im Tumormikromilieu. Vom Ausblick her betrachtet, eröffnet die Kartierung der dreidimensionalen Genomlandschaft in KrebsZellen neue Perspektiven für die Entwicklung zielgerichteter Therapien.
Durch die Identifikation von Enhancer-Rewirings oder mutationsbedingten regulatorischen Veränderungen lassen sich potenziell neue Biomarker erkennen und therapeutische Ansatzpunkte erschließen. Beispielsweise könnten maßgeschneiderte epigenetische Therapien entwickelt werden, die auf die Modulation der Enhancer-Promoter-Kommunikation abzielen. Auch die Charakterisierung von extrachromosomaler DNA als Quelle intensiver Genregulationsänderungen liefert Ansatzpunkte, um diese spezifischen DNA-Strukturen als Therapieziele ins Visier zu nehmen. Zusammenfassend lässt sich sagen, dass die dreidimensionale Genomorganisation eine zentrale Rolle bei der Regulation von Krebsgenen spielt und durch genetische und epigenetische Veränderungen im Tumorumfeld stark modifiziert wird. Die Kombination innovativer Methoden wie HiChIP und Einzelzellanalysen ermöglicht es, diese komplexen Zusammenhänge zu entschlüsseln und somit nicht nur das Verständnis der Krebsbiologie zu vertiefen, sondern auch die Grundlage für innovative Behandlungsmöglichkeiten zu schaffen.
Die Vernetzung von Struktur, Mutation und Funktion in der Genomlandschaft markiert dabei einen Meilenstein in der Onkologie und der personalisierten Medizin.


![Memetics – A Growth Industry in US Military Operations (2006) [pdf]](/images/E419C057-60DD-461C-8883-E541A693DF9A)