Die Erforschung unserer menschlichen Herkunft steht im Zentrum vieler genetischer Studien. Besonders spannend ist es, zurück in die Zeit zu blicken, als sich die ersten Populationen unseres modernen Menschen vom gemeinsamen Vorfahren abzweigten. Eine kürzlich veröffentlichte Studie hat mit Hilfe eines strukturierten koaleszenten Modells namens cobraa die komplexe Geschichte unserer Ahnen enthüllt und damit ein tieferes Verständnis der gemeinsamen Abstammung aller heutigen Menschen ermöglicht. Das neuartige Modell zeigt eine lange Periode der Aufspaltung und anschließendem Wiedervereinigung verschiedener Populationen, die fundamentale Auswirkungen auf die genetische Struktur unserer Spezies haben. Traditionelle Ansätze zur Analyse der menschlichen Populationen gingen oft von einer sogenannten panmiktischen Annahme aus – das bedeutet, dass die Populationen als ein großes, gemischtes Ganzes betrachtet wurden, in dem sich alle Individuen zufällig paaren und miteinander Gene austauschen.
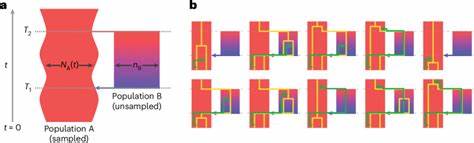
Doch genetische Daten der letzten Jahre haben diese Sichtweise zunehmend in Frage gestellt. Immer mehr Belege deuten darauf hin, dass es in der frühen Geschichte unserer Spezies wiederholt zu Phasen der Trennung und Vermischung, also Admixtur, gekommen ist. Das neue Modell greift dieses Thema gezielt auf, indem es explizit zwei auseinandergegangene, aber später wieder zusammengeführte Populationen berücksichtigt. Die Methode basiert auf einer coaleszenten Hidden-Markov-Modellierung, bei der genetische Sequenzen daraufhin analysiert werden, wie sich unterschiedliche Genlinien über die Zeit zurückverfolgen lassen. Anders als ältere Modelle, die nur die durchschnittliche Häufigkeit gemeinsamer Vorfahren betrachten, nutzt dieses Verfahren auch die Übergangswahrscheinlichkeiten zwischen verschiedenen coaleszenten Zeiten, was es erlaubt, komplexere historische Situationen wie Populationstruktur und Admixtur besser zu erfassen.
Anhand von genetischen Daten aus großen globalen Datenbanken wie dem 1000-Genomes-Projekt und dem Human Genome Diversity Project wurde das cobraa-Modell eingesetzt, um die menschliche Abstammungsgeschichte genauer zu rekonstruieren. Die Erkenntnis war überraschend und zugleich wegweisend: Unsere Vorfahren waren offenbar über eine lange Zeit hinweg in mindestens zwei Populationen geteilt, die sich vor etwa 1,5 Millionen Jahren voneinander trennten. Diese beiden Gruppen existierten für lange Zeit getrennt, bevor sie vor ungefähr 300.000 Jahren in einem Admixturereignis wieder zusammengeführt wurden. Bei diesem Vorgang floss etwa 80 Prozent der genetischen Informationen von der größeren Population A ein, während etwa 20 Prozent von der kleineren Population B stammten.
Eine detaillierte Analyse des Modells deutet zudem auf eine bedeutende Flaschenhalsphase in der größeren Population hin, die nach der Trennung von der kleineren Population entstand. Das heißt, die genetische Vielfalt in Population A wurde vorübergehend stark reduziert, was Auswirkungen auf die spätere genetische Struktur der modernen Menschen hatte. Außerdem konnten anhand der genomischen Daten Regionen identifiziert werden, die spezifisch von der Minderheitenpopulation B abgeleitet sind. Diese Regionen zeigen eine signifikante Korrelation mit der Nähe zu kodierenden Sequenzen im Genom und weisen darauf hin, dass genetische Varianten aus Population B vermutlich auf dem Hintergrund von Population A nachteilig wirkten und somit selektiv benachteiligt wurden. Die Verbindung dieser Erkenntnisse mit dem genetischen Abstand zu uralten Verwandten wie Neandertalern und Denisovanern ist besonders interessant.
Die Anteile der Mehrheitspopulation A zeigen eine starke Assoziation mit Divergenzen zu diesen archaischen Menschenarten, was darauf hindeutet, dass Population A auch der gemeinsame Vorfahre von Neandertalern und Denisovanern war. Dem gegenüber scheint der Beitrag von Population B genetisch deutlich weiter entfernt zu sein und spielte wohl eine untergeordnete Rolle in der Abstammung der archaischen Homininen. Das Modell wurde auch auf andere Tierarten angewandt, beispielsweise auf Delfine, Fledermäuse, Schimpansen und Gorillas. Die Ergebnisse unterscheiden sich dabei stark je nach Art und deren individuellen Evolutionsgeschichte. Während bei bestimmten Arten eine deutliche Struktur und alte Admixturereignisse erkannt wurden, zeigte sich bei anderen wenig bis keine Anzeichen für eine derart tiefgreifende Populationstruktur.
Der Nutzen des cobraa-Modells liegt unter anderem darin, dass es den zuvor bestehenden Identifizierbarkeitskonflikt löst, bei dem Unterschiedliche demographische Modelle anhand von durchschnittlichen Coaleszenzraten nicht mehr eindeutig unterschieden werden konnten. Durch die Berücksichtigung der Übergangswahrscheinlichkeiten zwischen benachbarten Koaleszenzzeiten wird zusätzliche Information erschlossen, die es ermöglicht, komplexere demographische Ereignisse mit besseren Parametern zu erfassen. Die Anwendung dieses Ansatzes brachte neue Perspektiven für die Diskussion über die menschliche Evolution. Die lange Zeitspanne der getrennten Populationen vor 1,5 Millionen Jahren fällt in eine Epoche, in der mehrere frühe Homo-Arten lebten, darunter Homo erectus und Homo heidelbergensis. Das Modell schlägt vor, dass diese frühen Populationen vielleicht als getrennte Linien bestanden, bevor sie sich wieder vermischten – ein Fakt, der bisher nicht ausführlich bestätigt werden konnte.
Dabei war Population A nicht nur die Hauptquelle des Genpools für heutige Menschen, sondern hat auch genetische Verbindungen zu den Archaischen wie Neandertalern und Denisovanern. Interessanterweise weist die Menge des genetischen Materials aus Population B von ungefähr 20 Prozent auf einen relativ hohen Anteil an Urbevölkerungshintergrund hin, der in allen heute lebenden Menschen zu finden ist. Dies ist viel höher als etwa die bekannten Anteile der archaischen Menschen in modernen Nicht-Afrikanern, die nur wenige Prozent betragen. Neben tieferen demographischen Erkenntnissen lassen sich auch genomische Regionen im Hinblick auf selektive Prozesse genauer untersuchen. So konnte gezeigt werden, dass gentechnische Materie aus der Minderheitenpopulation B wahrscheinlich Nachteile auf dem genetischen Hintergrund von Population A hatte, was durch Korrelationen mit funktionell wichtigen genetischen Regionen belegt wird.
Zugleich gibt es bereiche, in denen dieses Material überrepräsentiert ist, vor allem bei Genen, die mit neuronaler Entwicklung und Hirnfunktion zusammenhängen. Diese Beobachtung suggeriert, dass introgressives Material möglicherweise sogar adaptive Vorteile in speziellen biologischen Prozessen vermittelt hat. Die Studie bietet zudem ein mächtiges Werkzeug, um den Einfluss von struktureller Variation und alten Admixturen bei modernen Populationen besser zu verstehen. Dies kann entscheidend sein für die Interpretation von Genomdaten, die mit herkömmlichen Modellen oftmals schwer zu erklären sind. Kritische Aspekte der Methode sind allerdings die Annahmen eines plötzlichen Admixturereignisses ohne fortlaufenden Genfluss und die Annahme konstanter Mutations- und Rekombinationsraten.
In der Realität sind solche Parameter häufig komplexer und variabler. Dennoch zeigen Simulationen, dass diese Vereinfachungen keine wesentlichen Verzerrungen der Ergebnisse verursachen. Zukünftige Forschungsarbeiten könnten auf dieser Basis aufbauen, indem räumliche Variationen, mehrfach aufeinanderfolgende Admixturszenarien oder kontinuierlicher Genfluss explizit modelliert werden. Auch die Integration weiterer genetischer Informationen wie der Site-Frequency-Spectrum könnte die Auflösung und Genauigkeit der Modelle weiter verbessern. Überdies eröffnet das Modell neue Möglichkeiten, um präziser zu untersuchen, wie archaische Homininen in die Abstammungslinien der modernen Menschen eingebunden waren, insbesondere hinsichtlich der Frage, welchen Beitrag unterschiedliche archaische Populationen geleistet haben.






![Redis vs. Valkey Performance [video]](/images/B640AFA7-173C-44DB-9B92-50A0C9F8CC88)

