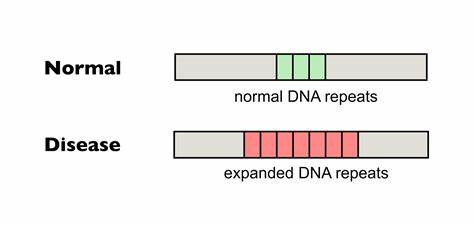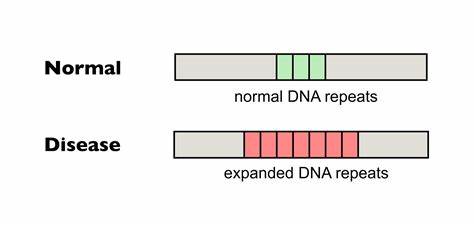
Die Huntington-Krankheit (HD) gehört zu den sogenannten trinukleotid-Repeat-Erkrankungen, die durch die pathologische Expansion von kurzen Wiederholungssequenzen im Erbgut verursacht werden. Speziell beruht HD auf einer verlängerten CAG-Trinukleotidsequenz innerhalb des HTT-Gens, die zu einem abnormalen Polyglutamin-Abschnitt im Huntingtin-Protein führt. Diese Erweiterung verursacht eine progressive, neurodegenerative Erkrankung mit unheilbarem Verlauf, die Bewegungsstörungen, kognitive Beeinträchtigungen und psychiatrische Symptome umfasst. Eine zentrale Herausforderung bei der Behandlung von HD ist die somatische Instabilität der CAG-Wiederholungen, das heißt deren fortschreitende Expansion in verschiedenen Körperzellen im Laufe des Lebens, insbesondere in Gehirnzellen, was die Krankheit verschlimmern und den Verlauf beschleunigen kann. Aktuelle Studien fokussieren sich darauf, wie sich diese somatischen Repeat-Expansionen durch gezielte genetische Interventionen verringern lassen.
Hochpräzise Gen-Editierungsmethoden wie die sogenannte Baseneditierung ermöglichen die gezielte Umwandlung einzelner Nukleotide innerhalb dieser Wiederholungssequenzen, wodurch deren Instabilität reduziert werden kann. Dabei werden sogenannte CAA-Unterbrechungen in die reine CAG-Sequenz eingefügt – eine Veränderung, die in der Natur als stabilisierend gilt. Synonyme Unterbrechungen wie CAA führen dazu, dass trotz der veränderten DNA-Sequenz weiterhin dieselbe Aminosäure Glutamin codiert wird, was bedeutet, dass die Funktion des Proteins Huntingtin unangetastet bleibt. Forschungen an Patientenzellen sowie an Mausmodellen der Huntington-Krankheit zeigten, dass die Anwendung von Cytosin-Baseneditoren (CBEs) zur Einführung dieser Unterbrechungen zu signifikant weniger somatischer Expansionen führt. In Zellkultur konnten relevante Fibroblasten von HD-Patienten effizient bearbeitet werden, wodurch bis zu 80 Prozent der Zellen nun eine stabilere CAG-Sequenz mit Unterbrechungen aufwiesen.
Langfristige Kultivierung dieser Zellen ergab, dass im Gegensatz zu unbehandelten Kontrollen keine weiteren schädlichen Wiederholungsexpansionen erfolgten. Dies deutet darauf hin, dass eine frühzeitige Intervention auf der DNA-Ebene das Fortschreiten der Krankheitsmechanismen wirksam hemmen kann. Ähnliche Erfolge wurden im Tiermodell, dem sogenannten Htt.Q111-Mausmodell, erzielt. Durch die neonatalen Injektionen von Adeno-assoziierten Viren (AAV), die Baseneditoren und passende Guide-RNAs transportieren, konnten in relevanten Hirnregionen wie dem Cortex und Striatum bis zu 70 Prozent der Huntingtin-Allele erfolgreich mit Unterbrechungen versehen werden.
Dies führte zu einer signifikanten Verringerung des Ausmaßes der CAG-Expansion im Gehirn. Zusätzlich ließen sich sogar erste Anzeichen einer Verkürzung der Repeat-Länge beobachten. Diese Resultate bestätigten in vivo die schützende Wirkung der Baseneditierung gegenüber der somatischen Expansion – einem Schlüsselaspekt bei der Krankheitsprogression. Die Bedeutung dieser Fortschritte besteht darin, dass die Stabilisierung oder Verkleinerung der CAG-Wiederholungen in neuronalem Gewebe eventuell eine Verzögerung des Erkrankungsbeginns bewirken oder das Fortschreiten der Symptome abmildern könnte. Schließlich beeinflusst die ursprüngliche Größe der CAG-Expansion bei Geburt zwar die Prognose, doch die dynamische somatische Instabilität trägt wesentlich zur Verschlechterung bei.
Deshalb eröffnet die Baseneditierung neue therapeutische Möglichkeiten, indem sie den genetischen Schaden nicht nur korrigiert, sondern auch die Tendenz zur weiteren Verschlechterung hemmt. Neben den HTT-CAG-Repeats konnte dieselbe Methodik auch bereits bei anderen trinukleotidischen Wiederholungserkrankungen wie der Friedreich-Ataxie angewendet werden. Dort kommen hingegen GAA-Expansions in einem anderen Gen vor, welche sich ebenfalls durch gezielte Unterbrechungen mit Adenin-Baseneditoren (ABEs) verarbeiten lassen. Diese ermutigenden Ergebnisse unterstreichen die Vielseitigkeit der Technik. Bei der Entwicklung solcher Therapien ist die Minimierung von Off-Target-Effekten ein entscheidender Faktor.
Durch umfassende Analysen mittels hochauflösender Ganzgenomsequenzierung und CIRCLE-seq konnten die potenziellen Nebenwirkungen der sgRNA-Bindung an andere repetitive oder ähnliche DNA-Sequenzen untersucht werden. Es zeigte sich, dass die meisten unerwünschten Änderungen entweder in nichtcodierenden Regionen des Genoms auftreten oder zu synonymeren Mutationen führen, die die Proteinfunktion nicht beeinträchtigen. Schwach ausgeprägte Nebenwirkungen konnten experimentell nachgewiesen werden, jedoch muss deren klinische Relevanz in zukünftigen Studien noch genau evaluiert werden. Insgesamt positioniert sich die Baseneditierung als vielversprechendes Werkzeug zur Therapie der Huntington-Krankheit und verwandter Erkrankungen mit trinukleotidischen Repeat-Expansionen. Die Fähigkeit, präzise eine stabile Variation in das verantwortliche Repeat-Genomsegment einzufügen, ohne das Protein selbst nachteilig zu verändern, bietet das Potenzial für innovative, krankheitsmodifizierende Therapiestrategien.
Die Forschung der letzten Jahre legt nahe, dass eine solche Gentherapie nicht nur in frühen Entwicklungsstadien, sondern auch nach Manifestation der Krankheit möglicherweise einen bedeutsamen Einfluss auf den Verlauf haben könnte. Weitere Herausforderungen bestehen jedoch in der Optimierung der Applikationsmethoden, um eine möglichst effiziente und spezifische Bearbeitung in den relevantesten Zelltypen zu gewährleisten. AAV-Mediatoren mit neuronaler Tropismus haben sich als wirksam erwiesen, doch alternative Vektoren und Verabreichungswege könnten den Zugang zu weiteren betroffenen Geweben wie Herz oder Gliazellen verbessern. Zudem sind die Langzeitfolgen der dauerhaften Baseneditor-Expression vor allem im klinischen Kontext noch nicht umfassend geklärt und müssen in präklinischen Modellen eingehend untersucht werden. Die neueste Datenlage zeugt von einem Paradigmenwechsel in der Behandlung monogener neurodegenerativer Erkrankungen: Durch das Eingreifen direkt auf der genetischen Bühne wird nicht nur symptomatisch behandelt, sondern die Ursache an der Wurzel adressiert.
Am Beispiel der Huntington-Krankheit zeigt sich, wie die Einführung natürlicher, nicht-pathogener Unterbrechungen in die CAG-Wiederholung die somatische Instabilität bremst und damit einen neuen therapeutischen Weg eröffnet. Diese Entwicklungen wecken Hoffnung für Betroffene und deren Familien und treiben die Forschung in Richtung gezielter, sicherer und nachhaltiger Genomeditierungen voran, die in Zukunft vielleicht das Leben von Menschen mit Huntington und anderen Repeat-Expansions-Erkrankungen maßgeblich verbessern können.