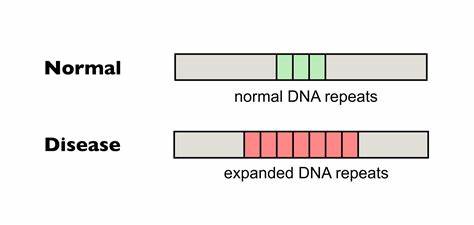
Die Huntington-Krankheit (HD) gehört zu den sogenannten trinukleotid-Repeat-Erkrankungen, bei denen sich kurze DNA-Sequenzen im Genom in krankheitsauslösender Weise wiederholen und erweitern. Konkret handelt es sich bei HD um eine Expansion der CAG-Trinukleotidwiederholung im HTT-Gen. Diese Repeats kodieren für Polyglutamin-Abschnitte im Huntingtin-Protein, die bei pathologischer Verlängerung neurodegenerative Prozesse auslösen und zum Funktionsverlust wichtiger Gehirnareale führen. Ein zentraler Aspekt bei HD ist die somatische Instabilität dieser Wiederholungen, also ihre fortschreitende Expansion in Zellen des Patientenlebens, insbesondere im zentralen Nervensystem. Dabei ist die Länge der CAG-Wiederholungen eng mit dem Krankheitsbeginn, Verlauf und der Schwere verknüpft.
Je länger die CAG-Kette ist, desto früher und gravierender manifestiert sich HD. Deshalb sind Ansätze, die somatische Repeat-Expansion einzudämmen, von großem Interesse. Bislang existieren keine kausalen Therapien, die die CAG-Repeat-Expansion direkt hemmen oder rückgängig machen können. Die klinische Behandlung fokussiert vorwiegend auf symptomatische Linderung, während Forschungsgruppen weltweit an genetischen und molekularen Interventionen arbeiten. In den letzten Jahren hat die Technologie der sogenannten Basen-Editierung große Aufmerksamkeit erlangt.
Diese präzise genetische Methode erlaubt es, einzelne Nukleotide im Erbgut gezielt umzuwandeln, ohne dass der DNA-Doppelstrang komplett zerschnitten werden muss, wie es bei der klassischen CRISPR/Cas9-Technologie oft der Fall ist. Die Basen-Editierung kann somit sicherer und kontrollierter Veränderungen einführen, die Mutationen korrigieren oder wie im Fall von trinukleotid-Repeat-Erkrankungen Modifikationen in repetitiven Sequenzen erzeugen. Forschende am Broad Institute und anderen Einrichtungen haben gezeigt, dass durch den gezielten Austausch einzelner Basen innerhalb der CAG-Repeats sogenannte Unterbrechungen eingefügt werden können. Typischerweise werden die repetitiven CAG-Sequenzen in gesunden Genomen durch Synonyme wie CAA unterbrochen, welche zwar auch Glutamin kodieren, aber die repetitiven Strukturen destabilisieren. Solche natürlichen Unterbrechungen stabilisieren das Genom und führen zu weniger somatischer Instabilität.
Das Fehlen dieser Unterbrechungen in den puren CAG-Trakten korreliert mit einer aggressiveren Form und einem früheren Auftreten von Huntington. Basierend auf dieser Erkenntnis verfolgen Wissenschaftler die Idee, gereinigte, pathologische CAG-Vervielfältigungen in HTT-codierenden Regionen mit Basen-Editoren so zu verändern, dass diese Unterbrechungen durch CAA-Triplets entstehen. In Zellkulturen von Huntington-Patienten konnten diese Basen-Editoren bereits zum Einsatz kommen. Die Forscher nutzen cytosinbasierte Editor-Varianten, die an cytosinreiche Bereiche binden und gezielt ein C in ein T umwandeln können (oder komplementär eine G-C-Basenpaarung in eine A•T-Basenpaarung überführen). Durch klug designte Guide-RNAs wurde der Basen-Editor zu den CAG-Regionen geleitet, um dort punktuelle Änderungen in der DNA durchzuführen.
Die Folge war eine markante Reduktion der somatischen Repeat-Expansion, das heißt, die CAG-Sequenzen verharrten stabiler in ihrer Länge und zeigten deutlich weniger Verlängerungen im Verlauf der Zellteilung. Diese Genomstabilisierung korrelierte mit einer reduzierten zellulären Schädigung und könnte deshalb entscheidend für die Verzögerung des Ausbruchs der neurodegenerativen Symptome sein. Ein ebenso wichtiger Aspekt war die Überprüfung potenzieller Off-Target-Effekte. Basen-Editoren könnten theoretisch auch an ähnlichen Sequenzen in anderen Gen-Regionen ansetzen und dort unbeabsichtigte Mutationen auslösen. Umfangreiche Analysen mit modernen Sequenzierungstechniken – darunter zirkulär amplifizierte DNA-Sequenzierung (CIRCLE-seq) und Whole Genome Sequencing – zeigten, dass die Off-Target-Aktivität zwar vorhanden, aber größtenteils in nicht-codierenden und damit weniger kritischen DNA-Bereichen zu finden war.
Außerdem wurden die meisten Änderungen als synonym oder neutral identifiziert, was positive Sicherheitsprofile für therapeutische Anwendungen eröffnet. Die erfolgreichen Experimente am Zellkulturmodell wurden anschließend in vivo in humanisierten Mausmodellen von Huntington mit pathologischen HTT CAG-Längen übertragen. Durch die Verabreichung von AAV9-Vektoren, welche die Basen-Editoren und die Target-gRNA enthielten, konnte in wesentlichen Gehirnregionen wie Kortex und Striatum eine effiziente Genommodifikation erzielt werden. Dort zeigten sich signifikante Reduktionen bei der somatischen Expansion der CAG-Repeats im Verlauf von mehreren Wochen bis Monaten sowie eine Erweiterung des genomischen Stabilitätsfensters. Die mouse-model-langszeituntersuchungen erbrachten zudem erste Hinweise darauf, dass diese Intervention auch somatische Kontraktionen der schädlichen CAG-Ketten begünstigt, was einen noch größeren therapeutischen Nutzen verspricht.
Dieses interdisziplinäre Forschungsprojekt zeigt eindrucksvoll, dass die Basen-Editierung eine neuartige Möglichkeit für die Behandlung von HD darstellt, indem sie direkt die genetische Ursache adressiert – nämlich die instabilen Trinukleotid-Repeat-Expansionen. Klinische Studien werden zukünftig klären müssen, ob sich dieser Effekt in menschlichen Nervenzellen ebenfalls realisieren lässt und ob die Behandlung neben der molekularen Stabilisierung auch den Verlauf der neurologischen Symptome günstig beeinflussen kann. Wichtig bei der Weiterentwicklung sind zudem technische Verbesserungen bei den Basen-Editoren, um die Effizienz weiter zu steigern und potenzielle Off-Target-Risiken weiter zu minimieren. Neue Varianten mit erhöhtem Target-Spezifitätsprofil und optimierte Delivery-Systeme mit gezieltem Nervenzell-Fokus könnten diese Vision untermauern. Darüber hinaus illustriert die Einführung von Unterbrechungen als therapeutisches Prinzip ein neues Paradigma im Umgang mit genetischen Wiederholungserkrankungen.
Es geht nicht zwingend nur um das Entfernen oder Korrigieren der Mutation, sondern vielmehr um die genetische Modifizierung von genetischen Strukturen in einer Weise, die deren pathogene Eigenschaften kompensiert oder abschwächt. Dies kann insbesondere bei schwierig zugänglichen oder komplexen Genloci wie den CAG-Trinukleotid-Repeat-Mutationen entscheidend sein. Parallel zu Huntington-Studien werden vergleichbare Basen-Editierungsansätze auch bei anderen Wiederholungserkrankungen wie der Friedreich-Ataxie oder SCA (Spinocerebelläre Ataxien) untersucht. Die gemeinsamen molekularen Mechanismen von Wiederholungsschleifen, DNA-Strukturinstabilitäten und DNA-Reparatur-Mechanismen bieten eine Grundlage zur breit angelegten Anwendung. Zusammenfassend eröffnet die präzise genetische Bearbeitung wiederholter DNA-Sequenzen mit Basen-Editoren Hoffnung auf erstmals ursächliche Therapien bei Huntington und verwandten genetischen Erkrankungen.
Die Verhinderung und Rückführung von somatischen Repeat-Expansionen konstituiert einen vielversprechenden Ansatz, der mit weiteren Fortschritten in der Geneditierungstechnologie zukünftig einen Weg zu veränderten Krankheitsverläufen und verbesserten Lebensqualität bieten kann.