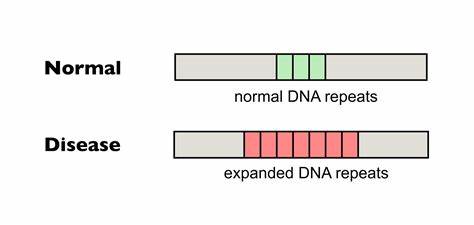
Die Huntington-Krankheit (HD) ist eine vererbte neurodegenerative Erkrankung, die durch die Expansion einer CAG-Trinukleotid-Wiederholung im HTT-Gen verursacht wird. Diese genetische Mutation führt zur Produktion eines mutierten Huntingtin-Proteins, das die Funktion von Neuronen beeinträchtigt und letztlich zu motorischen Störungen, kognitivem Abbau sowie psychiatrischen Symptomen führt. Einer der Hauptfaktoren für den Verlauf der Krankheit ist die Länge dieser CAG-Wiederholungen, die sich im Laufe des Lebens weiter ausdehnen können – ein Phänomen, das als somatische Repeat-Expansion bezeichnet wird. Neue Forschungen konzentrieren sich darauf, diese somatischen Expansionen zu reduzieren oder zu verhindern, um das Fortschreiten der Krankheit aufzuhalten oder zumindest zu verlangsamen. Ein besonders vielversprechender Ansatz ist die gezielte Genom-Editierung, die mithilfe von basenmodifizierenden Technologien Wiederholungsunterbrechungen einfügt und dadurch die Instabilität der tripletbasierten DNA-Sequenz vermindert.
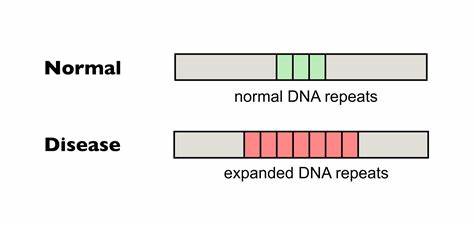
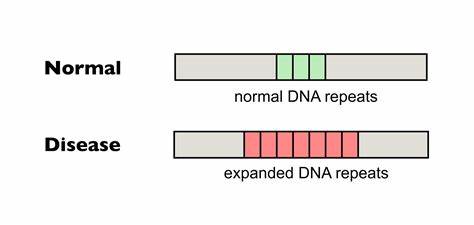
Im Kern der Huntington-Erkrankung steht die Instabilität der CAG-Wiederholungen, die im HTT-Gen als Polyglutamin-Abschnitt in der Huntingtin-Proteinkette abgebildet wird. Längere CAG-Trakte gelten als risikobehafteter und korrelieren mit früherem Erkrankungsbeginn und schwereren Symptomen. Doch nicht nur die Gesamtlänge, sondern vor allem die Reinheit der Wiederholungen beeinflusst die somatische Expansion. Natürliche Unterbrechungen in der CAG-Sequenz, beispielsweise durch die Einführung des ähnlichen Triplets CAA, können die Stabilität der DNA erhöhen. Diese Unterbrechungen vermindern die Neigung zur Bildung strukturierter DNA-Konformationen, wie sogenannten Haarpin-Schleifen, die oft als Auslöser für fehleranfällige DNA-Reparaturmechanismen dienen und so zur Expansion führen.
Die jüngsten Fortschritte in der Genom-Editierung ermöglichen es nun, diese natürlichen Unterbrechungen gezielt in den pathogenen Wiederholungstrakt einzufügen. Die sogenannte Base-Editing-Technologie erlaubt punktgenaue Einzelbasenveränderungen ohne die Notwendigkeit von DNA-Doppelstrangbrüchen. Spezielle cytosin- und adeninspezifische Enzyme, gekoppelt an eine modifizierte Cas9-Nuklease, können bestimmte Nukleotide in der DNA direkt verändern. Mit dieser Methode ist es möglich, CAG-Wiederholungen in CAA zu verwandeln, wodurch die Sequenzinterruption geschaffen wird und die Instabilität reduziert wird. In vitro-Studien an patienteneigenen Fibroblasten haben gezeigt, dass die Anwendung von cytosinbasierten Editoren zu einer signifikanten Einführung von CAA-Unterbrechungen im pathogenen HTT-Allelen führt.
Nach der Behandlung konnten bis zu 80 Prozent der Zellen Sequenzunterbrechungen aufweisen, was mit einer langfristigen Stabilisierung des CAG-Trakts einherging. Besonders bemerkenswert ist, dass über mehrere Zellkulturgenerationen hinweg die somatischen Expansionen deutlich reduziert wurden, wobei manche Trakte sogar eine Rückbildung der CAG-Länge erfuhren. Darüber hinaus wurden diese Editierungsmethoden auch erfolgreich in Mausmodellen der Huntington-Krankheit angewandt. Durch eine zielgerichtete AAV9-gestützte Abgabe der Base-Editoren in die zentrale Nervensystemregionen, wie den Kortex und Striatum, konnten Forscher wiederholte Einfügungen von CAA-Unterbrechungen beobachten. Dies ging einher mit einer signifikanten Verlangsamung der somatischen Expansion und einer Eindämmung der pathogenen Repeat-Länge.
Die Übertragung dieser Ergebnisse auf die Therapie menschlicher Patienten könnte die Grundlage für eine bahnbrechende Behandlung darstellen, die das Fortschreiten der Krankheit deutlich verzögern kann. Allerdings birgt die Genom-Editierung auch Herausforderungen. Die Off-Target-Aktivität, also das unbeabsichtigte Editieren anderer genomischer Stellen, muss sorgfältig überwacht und minimiert werden, um mögliche unerwünschte Mutationen zu verhindern. Die bisher durchgeführten Studien berichten, dass die meisten Off-Target-Effekte in nicht-kodierenden Regionen auftreten und größtenteils inharente Einzelbasenvarianten darstellen, die auch in der natürlichen Bevölkerung vorkommen. Dennoch ist eine umfassende Langzeitbeobachtung essentiell, vor allem bei therapeutischen Anwendungen im menschlichen Gehirn.
Die Komplexität der Wiederholungsinstabilität zeigt sich auch darin, dass neben der Länge des Repeat-Trakts auch Faktoren wie DNA-Repair-Mechanismen, Transkription und zellulärer Stoffwechsel maßgeblich Einfluss nehmen. Deshalb könnten Kombinationstherapien, die genomische Unterbrechungen mit pharmakologischen oder zellulären Interventionen kombinieren, zukünftig effektiver sein. Einige Medikamente, wie Omaveloxolon, sind bereits in der Behandlung anderer trinukleotidbasierter Krankheiten zugelassen und verbessern zumindest in Teilen die Symptomatik, stoppen die Grunderkrankung aber nicht. Technologisch eröffnen alternative Entwicklungen wie Prime Editing oder virale Vektorverbesserungen zusätzliche Möglichkeiten, um noch effizientere, präzise und sichere Editierungen bei differenzierten Zelltypen zu erreichen. Zudem bleibt die molekulare Charakterisierung der Huntington-Krankheit und der somatischen Instabilität im Fokus, um mögliche Biomarker für den Therapieerfolg oder die individuelle Prognose zu identifizieren.
Abschließend lässt sich zusammenfassen, dass die gezielte Base-Editing-Technologie einen revolutionären Ansatz darstellt, um das Fortschreiten der Huntington-Krankheit auf molekularer Ebene zu beeinflussen. Die Verringerung somatischer Repeat-Expansionen durch die Einführung genetischer Unterbrechungen in patienteneigenen Zellen und in Tiermodellen geht mit einer Stabilisierung des pathogenen HTT-Allels einher und eröffnet Perspektiven für zukünftige Therapien. Die erfolgreiche Applikation in vivo verdeutlicht das enorme Potenzial, das in der präzisen Genom-Editierung für neurodegenerative Erkrankungen liegt. Mit weiteren Fortschritten hinsichtlich Effizienz, Sicherheit und Auslieferung könnte diese Technologie eines Tages das Leben von Betroffenen nachhaltig verändern und den Kampf gegen Huntington entscheidend voranbringen.