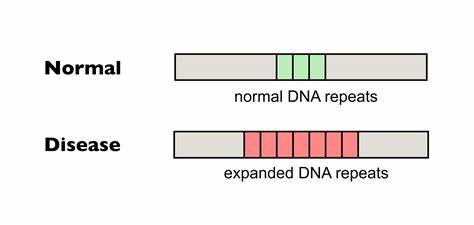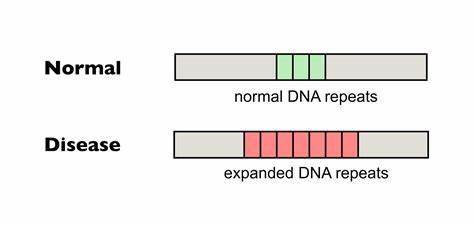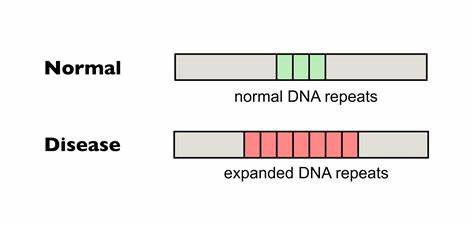
Huntington ist eine genetisch bedingte neurodegenerative Erkrankung, die durch die Expansion einer spezifischen Trinukleotid-Wiederholung im HTT-Gen verursacht wird. Diese Wiederholungen bestehen aus der Sequenz CAG. Ab einer bestimmten Länge wird der genetische Code instabil, was zur Vergrößerung der Wiederholungen in somatischen Zellen führt, ein Prozess, der als somatische Repeat-Expansion bezeichnet wird. Diese Expansionen korrelieren stark mit dem Auftreten und der Schwere der Krankheitssymptome und bilden damit einen zentralen therapeutischen Ansatzpunkt. Fortschrittliche Gen-Editing-Technologien bieten innovative Möglichkeiten, um diese pathologische Erweiterung zu verhindern oder zu verringern.
Insbesondere die präzise Einführung von Unterbrechungen innerhalb der CAG-Trinukleotid-Repeats ermöglicht es, die Instabilität der Wiederholungen zu reduzieren und somit die somatische Expansion zu bremsen. Forscherinnen und Forscher nutzen sogenannte Base Editing Verfahren, bei denen gezielte Punktmutationen ohne klassische DNA-Doppelstrangbrüche eingefügt werden können, um natürliche, nicht-pathogene Variationen nachzuahmen, die bei manchen Individuen eine stabilisierende Wirkung zeigen. Dabei kommt vor allem die Umwandlung von CAG zu CAA Codons zum Einsatz, die dieselbe Aminosäure Glutamin codieren, aber die Wiederholung strukturbiologisch unterbrechen und dadurch die Expansion verhindern. Studien in Patientenzellen und Mausmodellen haben gezeigt, dass solche Base Editing Methoden zu einer deutlichen Verringerung der somatischen CAG-Expansion führen können. Ein wichtiger Vorteil besteht darin, dass die genetische Information in den Zielgenen funktional erhalten bleibt, da die Aminosäuresequenz unberührt bleibt.
Das Verhindern oder Verlangsamen der somatischen CAG-Expansion stellt eine vielversprechende Strategie dar, um die Krankheitsprogression aufzuhalten oder zu verzögern, da insbesondere Nervenzellen, die von Huntington betroffen sind, lange Zeit ohne ausgeprägte Symptome fortlaufende Wiederholungsexpansionen akkumulieren. Sobald jedoch eine kritische Schwelle überschritten ist, setzt die Neurodegeneration ein. Die zeitnahe Intervention zur Stabilisierung der Repeat-Länge könnte somit eine immense klinische Bedeutung haben. Neben der direkten Base Editing-Technologie wurden auch innovative Vektorsysteme wie AAV9 genutzt, um die genetischen Werkzeuge gezielt in relevanten Gehirnregionen wie Cortex und Striatum abzugeben. Experimente mit der sogenannten Htt.
Q111-Maus, einem bewährten Modell der Huntington-Krankheit mit langem CAG-Repeat, belegten eine erfolgreiche Einführung von Unterbrechungen und eine signifikante Verringerung der somatischen Erweiterung in neuronalen Geweben. Die Effizienz der Editierung nahm mit der Zeit zu und blieb über Monate stabil, was für eine nachhaltige therapeutische Wirkung spricht. Zudem wurde dokumentiert, dass neben der Verhinderung der Expansion auch eine teilweise Kontraktion der bestehenden Repeat-Längen möglich ist, was einen zusätzlichen therapeutischen Vorteil darstellen kann. Neben Huntington zeigen Untersuchungen auch bei anderen trinukleotid-bedingten Erkrankungen, wie Friedreich-Ataxie, dass ähnliche Ansätze mit Adenin-Base-Editoren zur Interruptierung der pathogenen GAA-Wiederholungen erfolgreich sind und die somatische Expansion reduzieren. Die Forschungen liefern somit einen generalisierbaren Ansatz zur Behandlung von TNR-Erkrankungen.
Trotz der vielversprechenden Daten ist die Entwicklung von Gen-Therapien mit Base Editing noch mit Herausforderungen verbunden. So bestehen Risiken durch mögliche Off-Target-Effekte, die unerwünschte Veränderungen in anderen Genomelementen hervorrufen können. Um diese Risiken zu minimieren, wurden spezifische Cas9-Varianten mit erweitertem PAM-Spektrum und verbesserten Editier-Eigenschaften und Reinigungsverfahren eingesetzt. Erhobene Daten zeigen zudem, dass ein Großteil der Off-Target-Events entweder in nicht-codierenden Regionen auftritt oder synonyme Veränderungen bewirkt, die keine negativen funktionellen Konsequenzen haben. Dies untermauert die biologische Verträglichkeit der Methodik, erfordert jedoch weitere präklinische Sicherheitsstudien, bevor klinische Anwendungen möglich sind.
Die Forschung verbindet modernste Molekularbiologie, Genomik und Neurobiologie, um eine bisher unabdingbare therapeutische Lücke zu schließen. Während derzeitige Therapieoptionen für Huntington hauptsächlich Symptome mildern und keine kausale Behandlung bieten, erlauben die jüngsten Fortschritte bei der präzisen Gen-Editierung einen zielgerichteten Eingriff im Genmaterial eines Patienten. Dies könnte in Zukunft die Krankheitsausprägung nicht nur verzögern, sondern eventuell auch aufhalten. Die Basis hierfür bilden langjährige Erkenntnisse zur molekularen Pathogenese der Repeat-Expansionen, kombiniert mit der Innovation durch die CRISPR-basierten Base Editing Technologien. Darüber hinaus stärken die gewonnenen Erkenntnisse zum Einfluss von Repeat-Interruptions auf die somatische Instabilität das Verständnis der Krankheitsprogression und eröffnen neue diagnostische und therapeutische Perspektiven.
Insbesondere die Verkürzung der somatischen Expansion oder deren Stabilisierung sollte das Auftreten von Symptomen hinauszögern, was Patienten eine bessere Lebensqualität und häufig eine verlängerte phänotypische Latenz bietet. Trotz des großen Potenzials, sind noch weitere Schritte nötig, um Effizienz, Sicherheit und zielgerichtete Verteilung in menschlichen Geweben zu optimieren. Hierzu zählen auch alternative Vektorsysteme, Verabreichungswege sowie eine fein abgestimmte Kontrolle der Expression von Base Editors. Die Umsetzung in klinische Studien wird die nächsten Jahre prägen. Insgesamt bietet der aktuelle Forschungsstand einen hoffnungsvollen Einblick in die Zukunft der Behandlung von Huntington und verwandten trinukleotidbedingten Erkrankungen.
Durch die gezielte Bearbeitung von Wiederholungssequenzen, die das zentrale molekulare Problem darstellen, eröffnen sich neue therapeutische Strategien, die das Fortschreiten neurodegenerativer Erkrankungen signifikant bremsen könnten. Diese Entwicklungen markieren einen Paradigmenwechsel in der personalisierten Genommedizin und können die Prognose für viele Patientinnen und Patienten weltweit verbessern.