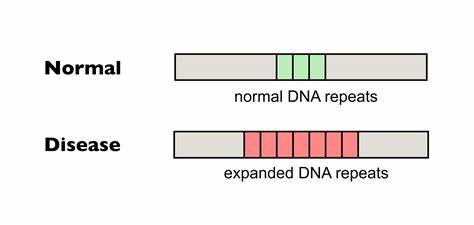
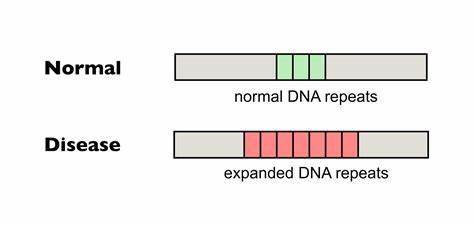
Huntington ist eine erbliche neurodegenerative Erkrankung, die durch eine pathologische Expansion von CAG-Trinukleotid-Wiederholungen im HTT-Gen verursacht wird. Diese Wiederholungen bestehen aus der Aneinanderreihung von Cytosin, Adenin und Guanin in der DNA-Sequenz und kodieren für eine glutaminreiche Aminosäurekette im Huntingtin-Protein. Die Länge dieses CAG-Traktes spielt eine zentrale Rolle für den Krankheitsbeginn und die Schwere der Symptome. Eine Besonderheit dieser Erkrankung ist jedoch nicht nur die genetische Ausgangslage bei Geburt, sondern auch die sogenannte somatische Expansion, bei der sich die Anzahl der CAG-Wiederholungen im Laufe des Lebens in verschiedenen Geweben, insbesondere im zentralen Nervensystem, weiter vermehren kann. Diese Heterogenität in der Zellanordnung und die zunehmende Anzahl der Wiederholungen in ausgewählten Zellen stehen im Zusammenhang mit einem beschleunigten Ausbruch und einer schnelleren Progression der Erkrankung.
Die Erfassung und Regulation dieser somatischen Instabilität ist daher ein zentrales Anliegen der Huntington-Forschung. In jüngster Zeit führte die Anwendung von Baseediting-Technologien zu bedeutenden Fortschritten. Im Gegensatz zu klassischen Genscheren, die DNA-Doppelstrangbrüche erzeugen, erlauben Baseeditoren die gezielte Änderung einzelner Nukleotide ohne die DNA vollständig zu schneiden. Dies bietet die Möglichkeit, die CAG-Wiederholungen durch gezielte, sogenannte synonyme Interruptionsmutationen mehr stabiles Sequenzmaterial einzufügen, ohne die kodierte Aminosäureabfolge des Huntingtin-Proteins zu verändern. Konkret bedeutet dies, dass durch die Umwandlung bestimmter CAG-Codons in CAA-Codons ein stabileres, sogenanntes „unterbrochenes“ Repeat-Muster entsteht, das weniger anfällig für somatische Instabilität ist.
Forschende konnten zeigen, dass natürliche Variationen mit solchen Interruptionssequenzen bei Patienten mit längeren CAG-Trakten ein verzögertes Krankheitsalter sowie eine geringere somatische Expansion aufweisen. Dies lieferte die initiale Hypothese, dass das gezielte Einfügen solcher Interruptionsmutationen mittels Baseeditoren bei Huntington-Patientenzellen die somatische Somatische Repeat-Expansion vermindern und damit die neurodegenerative Krankheitsprogression potenziell verlangsamen kann. Zahlreiche Studien, darunter präklinische Versuche mit patientengewonnenen Fibroblasten und neurodegenerativ relevanten Mausmodellen, bestätigten diese Annahme. So wurden effektive Cytosin-Baseeditoren eingesetzt, um die CAG-Repeats im HTT-Gen gezielt an den entsprechenden DNA-Stellen in neuronalen Zellen zu verändern. Mit Hilfe von AAV9-Vektoren gelang die effiziente Übertragung des Baseediting-Systems in die Hirngewebe von Mäusen mit einem Huntington-Modell, das menschliche HTT-Repeats mit pathologischer Länge bei physiologischer Expression aufwies.
Die Ergebnisse dieser In-vivo-Studien sind ermutigend. Bereits nach wenigen Wochen konnte eine signifikante Anzahl von HTT-Allelen mit der gewünschten CAA-Unterbrechung nachgewiesen werden. Diese Modifikation führte zu einer messbaren Reduktion der somatischen Expansion des Repeat-Traktes in lebenswichtigen Hirnregionen wie dem Kortex und Striatum. Von besonderer Bedeutung war die Feststellung, dass der CAG-Trakt nicht nur in der Expansion gehemmt wurde, sondern sogar teilweise wieder verkürzt werden konnte. Diese Kontraktionen zeigen, dass die Genom-Editierung nicht nur ein Halt voranschreitender Kürzungen und struktureller Veränderungen darstellt, sondern auch zum potenziellen Schrumpfen pathologisch verlängerter Repeat-Trakte beitragen kann.
Neben der Effizienz und Wirksamkeit ist bei der Anwendung von Baseeditoren auch die Spezifität entscheidend. Da CAG-Wiederholungen in verschiedenen Genomregionen vorkommen, war eine genaue Analyse der Off-Target-Effekte erforderlich. Experimentelle Genome-weite Off-Target-Analysen zeigten überwiegend Availität der Editierung an den eigentlichen Zielorten mit minimalen Nebenwirkungen. Bisherige Studien konnten wichtige off-target-bezogene Indel-Mutationen oder potentiell schädlichen Aminosäureveränderungen entweder nicht oder nur in äußerst geringem Umfang nachweisen. Dieser Aspekt unterstreicht die Sicherheitspotentiale der Baseediting-Technologie im therapeutischen Kontext.
Darüber hinaus zeigen aktuelle Untersuchungen, dass der therapeutische Nutzen in der Verhinderung der somatischen Instabilität überwiegt. Denn die Expansion der CAG-Trakte im zentralen Nervensystem wird als maßgeblicher Faktor für Krankheitsbeginn und -progression angesehen. Die Basis-Editor-gesteuerte Unterbrechung der puren CAG-Trakte erhöht die genetische Stabilität und kann den Schwellenwert für pathologische Effekte im Verlauf hinauszögern. Neben den bisherigen Anwendungsgebieten ist bei Huntington auch die Frage relevant, ob eine frühe Intervention im neonatalen oder frühen postnatalen Alter notwendig ist, um effektiv die Somatische Instabilität zu bekämpfen. Studien mit Mausmodellen verweisen darauf, dass ein frühzeitiges Treatment eine bessere Verhinderung der Expansion ermöglicht und somit die neurodegenerative Entwicklung langsamer erfolgt.
Es besteht jedoch noch Forschungsbedarf, ob spätere, gleichwohl effektive Editierungen ebenfalls therapeutisch wirksam sind. Die Entwicklung dieser Technologien und ihre Weiterentwicklung zur klinischen Anwendung zeigen einen vielversprechenden Weg, die genetische Ursache sowie den Verlauf der Huntington-Erkrankung an der molekularen Basis anzugehen. Die Vermeidung oder Reduktion somatischer Repeat-Expansionen könnte nicht nur die Auslösung der Erkrankung hinauszögern, sondern auch die Gesamtdauer und Schwere der Symptome signifikant vermindern. Diese Innovation bringt große Hoffnung für Patienten, teilweise vorhandene genetische Risiken durch präzise Baseediting-Technologien zu minimieren. Die Herausforderung wird sein, die effiziente und sichere Lieferung der Editoren in humanes Gewebe zu gewährleisten und mögliche immunologische oder unerwünschte molekulare Nebenwirkungen zu vermeiden.
Zukünftige Studien werden sich daher auf Langzeitwirkungen, die Wirksamkeit im Erwachsenenalter und die Robustheit der Baseediting-Systeme konzentrieren müssen. Zudem gilt es, Verabreichungsstrategien, etwa über Einmalimpfungen mit Adeno-assoziierten Viren oder transienten Editoren, zu optimieren. Die Kombination mit anderen therapeutischen Ansätzen könnte ebenfalls Synergien hervorbringen. Schließlich unterstreicht die Basiseditor-getriebene Interruptions-Strategie das Potenzial genetischer Modifikation, gezielt krankheitsauslösende Mutationen subtil und genau mit minimalem Risiko zu korrigieren. Angesichts der schweren und bislang unheilbaren Natur der Huntington-Krankheit stellt dies einen Meilenstein in der molekularen Neurowissenschaft und Medizin dar.
Die spannende Verbindung von präziser Genom-Editierung, besserem Verständnis der molekularen Pathomechanismen und innovativen Therapiekonzepten verspricht, Huntington in eine behandelbare Krankheit zu verwandeln – und somatische Repeat-Expansionen zu einem steuerbaren Faktor im Kampf gegen neurodegenerative Erkrankungen zu machen.




!['Maybe Venice is the city that can save the world' [video]](/images/471FB54D-C379-4E1E-B2B4-1AC8D11C9644)