Huntington ist eine schwerwiegende neurodegenerative Erkrankung, deren Ursache in der krankhaften Ausweitung bestimmter DNA-Wiederholungen, sogenannter Trinukleotid-Repeats, im HTT-Gen liegt. Diese Wiederholungen, vor allem die Sequenz CAG, kodieren für die Aminosäure Glutamin und führen bei pathologischer Ausdehnung zur Produktion eines toxischen Proteins mit verlängerten Polyglutamin-Strecken. Die Folge ist ein fortschreitender Verlust neuronaler Funktion, der sich klinisch in motorischen Störungen, kognitivem Abbau und psychiatrischen Symptomen manifestiert. Trotz intensiver Forschung existiert bislang keine kurative Therapie, die den Krankheitsverlauf signifikant aufhalten kann. Die instabile Natur dieser Repeat-Sequenzen innerhalb der somatischen Zellen verschärft die Problematik zusätzlich, da sie sich im Laufe des Lebens ausweiten und somit die Krankheitsschwere verstärken können.
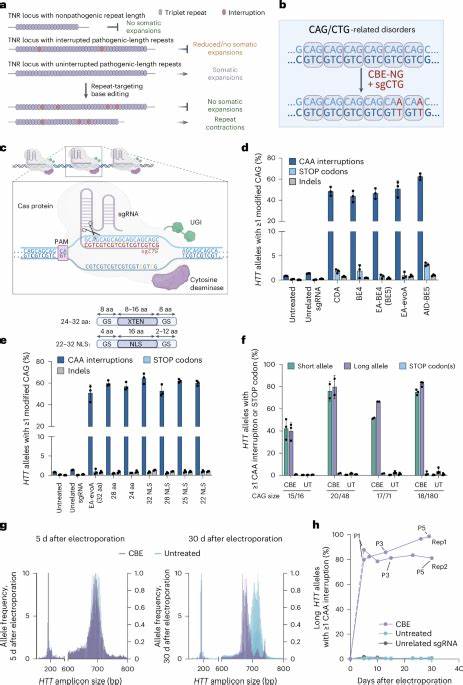
Die Dynamik dieser Repeat-Erweiterungen wurde lange Zeit als Hindernis betrachtet. Somatische Instabilität bedeutet, dass die Anzahl der CAG-Wiederholungen in verschiedenen Geweben und Zellen unterschiedlich ist und mit der Zeit tendenziell zunimmt. Besonders betroffen sind Hirnregionen, die bei Huntington degenerieren, wie der Striatum. Diese Weiterentwicklung der Expansion ist ein zentraler Faktor für den Krankheitsfortschritt und gilt als potenzielles therapeutisches Ziel. Eine neuartige Herangehensweise nutzt die Möglichkeiten der Basen-Editierung, einer Form der präzisen Genom-Editierung, die einzelne Nukleotide markiert und umwandelt, ohne die DNA-Doppelhelix zu durchtrennen.
Dabei kommen Enzyme zum Einsatz, die gezielt Cytosin in Thymin oder Adenin in Guanin umwandeln können. Durch die gezielte Einfügung von sogenannten Interruptions innerhalb der CAG-Repeats – das heißt, kleine Veränderungen der Nukleotidsequenz, welche aber die kodierten Aminosäuren nicht verändern – wird die Homogenität der Repeat-Sequenz unterbrochen. Solche Unterbrechungen sind in der Natur bekannt und mit einer stabileren DNA-Sequenz sowie einem verzögerten Krankheitsbeginn assoziiert. In aktuellen Studien konnte gezeigt werden, dass durch Cytosin-Basen-Editoren in Kombination mit spezifischen leitenden RNA-Molekülen (sgRNA), sogenannte CAG-CBEs, Interrupted CAA-Codons in den HTT-Repeats eingefügt werden können. Diese Änderung ist synonym, bedeutet also, dass die Aminosäure Glutamin unverändert bleibt.
Die Folge ist jedoch eine Reduktion der somatischen Instabilität. In Patienten-Zelllinien wurden nach der Behandlung mit dieser Methode eine deutliche Reduktion der Expansion von CAG-Repeats beobachtet. Es konnte sogar eine leichte Kontraktion, also eine Verkürzung der Repeat-Länge, festgestellt werden, was eine therapeutisch bedeutende Wirkung andeutet. Parallel dazu wurden in Mausmodellen, die das menschliche HTT-Gen mit pathologischer Repeat-Länge tragen, Adeno-assoziierte Viren (AAV) eingesetzt, um die Basen-Editoren gezielt in Hirngewebe einzubringen. So konnte die Zusammensetzung der HTT-Repeat-Sequenz verändert und sogenannte Interruptions eingebaut werden, was die somatische Expansion in relevanten Hirnarealen wie Cortex und Striatum signifikant verminderte.
Damit wurde ein Nachweis erbracht, dass die somatische Expansion zumindest teilweise durch diese gezielten, synonymer Codon-Unterbrechungen kontrollierbar ist. Diese Erkenntnisse weisen auf ein vielversprechendes therapeutisches Potenzial für Huntington und möglicherweise weitere Trinukleotid-Repeat-Erkrankungen mit ähnlicher Pathophysiologie hin. Besonders interessant ist das mechanistische Verständnis hinter der Stabilisierung der Repeat-Sequenz durch Interruptions. Die ursprünglichen CAG-Homopolymerstrecken neigen dazu, komplexe sekundäre DNA-Strukturen wie Haarnadeln oder R-Loops zu bilden, welche wiederum Fehler bei der DNA-Replikation und Reparatur begünstigen und zur Expansion führen. Das Einfügen von CAA oder anderen Unterbrechungscodons verhindert die Ausbildung dieser stabilen Fehlstrukturen und reduziert somit die Wahrscheinlichkeit von Repeat-Expansionen.
Neben Huntington betrifft dieses Prinzip auch andere neurodegenerative Erkrankungen, insbesondere Friedreich-Ataxie, bei der GAA-Repeats im FXN-Gen verlängert sind. Ähnlich wie bei den CAG-Repeats konnten durch Adenin-Basen-Editoren (ABEs) Interruptions innerhalb der GAA-Sequenzen eingefügt werden, was ebenfalls zu einer Reduktion der somatischen Instabilität führte und die Expression des FXN-Gens teilweise wiederherstellte. Auch hier wurden positive Ergebnisse in patienten-abgeleiteten Zellkulturen sowie in Mausmodellen erzielt, was die universelle Anwendbarkeit der Methode unterstreicht. Die Herausforderung bei basenbasierten Repeat-Editoren liegt im präzisen und effizienten Targeting der oft langen und hochrepetitiven Sequenzen, die auch an vielen anderen Stellen im Genom vorkommen. Durch die Verwendung von Cas9-Varianten mit erweitertem PAM-Spektrum und hochspezifischen sgRNAs konnte eine ausreichende Zielgenauigkeit erzielt werden.
Trotzdem ist die sorgfältige Evaluierung von Off-Target-Effekten unverzichtbar, um unerwünschte genetische Veränderungen an anderen genomischen Stellen auszuschließen. Moderne Off-Target-Nominierungsmethoden wie CIRCLE-seq und hochauflösende Ganzgenomsequenzierungen haben gezeigt, dass das Ausmaß solcher Nebenwirkungen gering und überwiegend in nichtkodierenden Bereichen lokalisiert ist. Synonyme und bekanntermaßen nicht schädliche Mutationen treten häufiger auf als deleterische Veränderungen. Der Einsatz von AAV9 als Vektor in den tierexperimentellen Studien bietet eine neuronalspezifische Transduktion und ermöglicht eine effiziente und anhaltende Expression der Basen-Editoren. Durch die Anwendung von neonatalen Injektionsverfahren konnte die frühe Intervention demonstriert werden, die einen erheblichen Einfluss auf die Verhinderung der Expansion bis ins Erwachsenenalter hat.
Langfristige Studien sind notwendig, um den möglichen Effekt auf Krankheitsprogression, neuronalem Zelltod und Verhaltensparametern zu evaluieren. Aus klinischer Sicht eröffnet die Möglichkeit, mittels Basen-Editing gezielt und dauerhaft die genetische Ursache der somatischen Repeat-Instabilität zu adressieren, eine ganz neue Generation von Therapien für Huntington und verwandte Erkrankungen. Während derzeitige Ansätze vorwiegend auf Symptombehandlung und teilweise auf die Reduktion der Gesamtgenexpression abzielen, bietet die gezielte Modifikation der Repeat-Struktur eine subtile, aber effektive Strategie, die das Risiko von Nebenwirkungen durch Proteinmangel minimiert. Ein wichtiger Aspekt für die zukünftige Entwicklung hin zur klinischen Anwendung ist die Verbesserung der Liefermethoden, Minimierung der Off-Target-Effekte und Optimierung der Editor-Effizienz. Die Kombination mit transienter Expression, zelltypspezifischen Promotoren und möglichen nicht-viralen Lieferwegen wie viralen Partikeln oder nanoparticlebasierten Ansätzen könnte die Sicherheit erhöhen.
Zusammenfassend lässt sich festhalten, dass die gezielte Basen-Editierung von Trinukleotid-Repeats eine wegweisende Methode darstellt, um die somatische Expansion der Repeat-Längen zu reduzieren und somit das Fortschreiten von Huntington und verwandten Erkrankungen abzubremsen. Die wissenschaftliche Evidenz aus zellulären und tierexperimentellen Modellen liefert eine solide Grundlage für weitere Forschungs- und Entwicklungsarbeiten mit Potenzial, die Lebensqualität von Patienten nachhaltig zu verbessern und – im Idealfall – künftig eine Heilung zu ermöglichen.