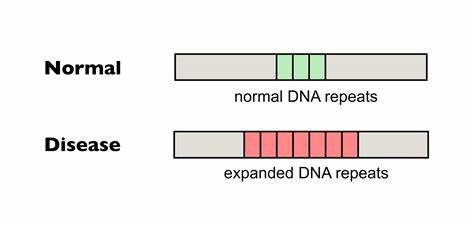
Die Huntington-Krankheit (HD) zählt zu den erblichen neurodegenerativen Erkrankungen, die durch eine pathologische Expansion von CAG-Trinukleotidwiederholungen im HTT-Gen ausgelöst wird. Diese ungewöhnlich langen Wiederholungen führen zur Synthese eines toxischen Proteins mit verlängerten Polyglutaminsequenzen, welche Nervenzellen insbesondere in bestimmten Hirnregionen schädigt und letztlich zum fortschreitenden Funktionsverlust führt. Ein charakteristisches Merkmal der Erkrankung ist die Instabilität und weitere Ausdehnung des CAG-Repeats in somatischen Geweben während des Lebens des Patienten, was als somatische Expansion bezeichnet wird. Dieses Phänomen korreliert stark mit dem Krankheitsbeginn und dem Verlauf, sodass ein größerer somatischer Repeat-Expansion eine schnellere Neurodegeneration fördert. Die bisher vorhandenen Therapien bei Huntington konzentrieren sich hauptsächlich auf die Linderung von Symptomen, bieten jedoch keine Möglichkeit, die Ursache der Krankheit zu bekämpfen oder den Prozess der somatischen Expansion aufzuhalten.
In diesem Zusammenhang eröffnet die fortschrittliche Präzisionsgenomeditierung, insbesondere das sogenannte Basen-Editing, neue Perspektiven. Es handelt sich dabei um eine Technologie, die gezielte, punktuelle Veränderungen einzelner Basenpaare in der DNA erlaubt, ohne Doppelstrangbrüche hervorzurufen, wie dies beispielsweise bei klassischen CRISPR-Cas9-Systemen der Fall ist. Forscher haben sich darauf konzentriert, durch Basen-Editing gezielt kleine Veränderungen innerhalb der CAG-Repeat-Abschnitte einzuführen, um deren Struktur zu destabilisieren und somit die somatische Instabilität zu reduzieren. Dabei werden beispielsweise CAG-Wiederholungen durch sogenannte Interruptions—begrenzt eingestreute Veränderungen wie CAA-Codons—unterbrochen, die auch natürlich bei manchen Menschen vorkommen und mit einem milderen Krankheitsverlauf sowie weniger aggressiver Repeat-Expansion assoziiert sind. Im Labor wurde eine Vielzahl an cytosin- und adenindezaminierenden Baseneditoren erprobt, die spezifisch die DNA sequenziell innerhalb des repetitiven Trinukleotidmusters modifizieren können.
Diese Enzyme werden über virale Vektoren, darunter häufig das Adeno-assoziierte Virus (AAV), effizient in Patientenzellen und experimentell in Mäusen eingebracht. Die baseneditierten Veränderungen führen zu sogenannten synonymen Interruptions-Codons, die am Proteinprodukt nichts verändern, aber die DNA-Struktur stabilisieren und die Neigung zur weiteren Expansion senken. Die wissenschaftlichen Ergebnisse zeigen, dass die gezielte Einführung dieser Interruptions in der HTT-Gensequenz von Fibroblasten, welche von Huntington-Patienten stammen, deutlich die Entstehung weiterer somatischer Repeat-Expansionen verlangsamt oder sogar verhindert. Dabei ist die Editierung besonders effizient in den lang expandierten pathogenen Allelen, was den therapeutischen Nutzen dieser Methode verstärkt. Darüber hinaus bestätigen tierexperimentelle Studien in Mäusen, die menschliche HTT-Gensequenzen mit pathologischen Repeat-Längen tragen, dass bereits eine moderate Editierquote im Gehirn, insbesondere in den für die HD relevanten Regionen wie Cortex und Striatum, die somatische Expansion im Gewebe signifikant vermindert.
Besonders hervorzuheben ist, dass diese Intervention auch zu so genannten Repeat-Kontraktionen führen kann, also zur verkürzten Wiederholungszahl, was einen zusätzlichen Schutz gegen die Krankheitsprogression bietet. Die langfristige Expression der Baseneditoren vermittelt eine kumulative Ansammlung dieser Interruptions und hält die Repetition stabil unterhalb eines kritischen Schwellenwerts, der mit neurodegenerativem Schaden verknüpft ist. Ein weiterer Vorteil dieser Strategie ist die hohe Spezifität der Editierung. Umfangreiche Analysen von Off-Target-Effekten, welche die unerwünschten Veränderungen an anderen genomischen Stellen beschreiben, ergeben, dass die meisten Veränderungen auf repetitive, nicht krankheitsbezogene Regionen begrenzt bleiben. Nur eine geringe Anzahl von Protein-codierenden Genen wird mit synonymen oder biologisch wenig relevanten Änderungen betroffen, was die Sicherheit einer solchen Therapie unterstreicht.
Dies ist ein bedeutender Fortschritt angesichts der bisherigen Herausforderungen bei der Genom-Editierung mit konventionellen Verfahren. Neben Huntington ist auch die Friedreich-Ataxie (FRDA) ein Beispiel für eine von Trinukleotid-Wiederholungen verursachte Erkrankung, hier zumeist ausgelöst durch die Expansion von GAA-Repeat-Abschnitten im FXN-Gen. Analog zu den CAG-Interruptions im Huntingtin-Gen hat die gezielte Einführung von A•T zu G•C Basesubstitutionen in den GAA-Abschnitten die somatische Instabilität reduziert und die FXN-Expression verbessert, was die zellulären Defekte abschwächt. Die Erfolge bei FRDA deuten darauf hin, dass das Basen-Editing an Trinukleotid-Wiederholungen eine allgemeine therapeutische Methode für verschiedene neurodegenerative Trinukleotid-Erkrankungen sein könnte. Die Herausforderung besteht jedoch darin, die effiziente, sichere und zielgerichtete Auslieferung der Baseneditoren in den menschlichen Organismus zu gewährleisten.
Neonatale intrazerebroventrikuläre Injektionen in Mausmodellen stellen zwar einen machbaren Weg dar, doch bei Menschen bedarf es noch weiterer Innovationen hinsichtlich Delivery-Systemen, Gewebespezifität und Transientität des Editors. Fortschritte in der Vektor-Technologie, temorärer Expression und gezieltem Transport könnten diese Therapie zukünftig praktikabel machen. Insgesamt markieren die jüngsten Forschungsergebnisse einen bedeutenden Schritt in Richtung gezielter therapeutischer Eingriffe bei Huntington und verwandten Trinukleotid-Expansionserkrankungen. Die Möglichkeit, somatische Repeat-Expansionen durch gezielte Baseneditierung zu bremsen oder gar umzukehren, releast nicht nur neue Hoffnung für Betroffene, sondern fordert die Neuinterpretation des genomischen Managements von instabilen Repeat-Sequenzen heraus. Zukünftige Studien werden sich darauf konzentrieren, langfristige Sicherheit, optimale Editiergewebe, Auslieferungswege sowie die potenzielle Kombination mit anderen Therapien wie Antisense-Oligonukleotiden oder Small-Molecule-Ansätzen zu evaluieren.
Eine verbesserte Kenntnis der molekularen Mechanismen, welche die somatische Instabilität steuern, wird darüber hinaus dazu beitragen, weitere Zielstrukturen für die Modulation zu identifizieren. Für Patienten mit Huntington bietet das Editieren von CAG-Wiederholungen somit eine innovative Möglichkeit, direkt an der Ursache der Krankheit anzusetzen. Die gezielte Stabilisierung der Repeat-Sequenz durch Interruptions könnte die fatalen somatischen Expansionen verhindern, die für die Progression der Erkrankung mitverantwortlich sind. Während klinische Anwendungen noch in den Kinderschuhen stecken, ist die Richtung klar: Die Genombearbeitung eröffnet neue Wege in der Behandlung bislang unheilbarer neurodegenerativer Erkrankungen.