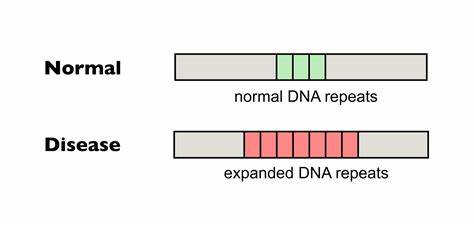
Huntington-Krankheit (HD) ist eine verheerende neurodegenerative Erkrankung, die durch eine Expansion von CAG-Trinukleotidwiederholungen im Huntingtin-Gen (HTT) verursacht wird. Diese genetische Anomalie führt zur Produktion eines defekten Proteins, das in Nervenzellen toxisch wirkt und letztlich zu Symptomen wie unkontrollierten Bewegungen, kognitiven Beeinträchtigungen und psychiatrischen Störungen führt. Die Grundlage des Krankheitsverlaufs ist dabei entscheidend von der Länge und Stabilität dieser CAG-Wiederholungen abhängig. Ein besonders wichtiger Mechanismus zur Verschlechterung der Krankheit ist die somatische Expansion der Wiederholungen in bestimmten Geweben, vor allem im zentralen Nervensystem. Je länger die CAG-Repeats in Neuronen werden, desto stärker setzt der molekulare Schaden ein und desto früher und gravierender sind die Symptome.
Die Verringerung oder Verhinderung dieser somatischen Expansionen stellt daher einen vielversprechenden Ansatz für neue Behandlungsmöglichkeiten dar. Traditionelle Therapieansätze bei Huntington konzentrieren sich häufig auf die Linderung von Symptomen, doch echte kausale Interventionen sind bisher nicht etabliert. In den letzten Jahren hat die genetische Forschung jedoch bedeutende Fortschritte gemacht, insbesondere im Bereich der präzisen Genom-Editierung. Aktuelle Studien haben gezeigt, dass gezielte Bearbeitung der trinukleotidischen Wiederholungsregionen im HTT-Gen, insbesondere durch sogenannte Base-Editing-Technologien, die Stabilität der Wiederholungssequenzen verbessern kann. Diese Methode nutzt modifizierte CRISPR/Cas-Systeme, die einzelne Basen der DNA direkt umwandeln, ohne Doppelstrangbrüche zu verursachen.
Durch die Einführung natürlicher Unterbrechungen in der CAG-Sequenz, beispielsweise durch CAA-Codons, die die gleiche Aminosäure (Glutamin) kodieren, lassen sich die Wiederholungen stabilisieren und die Neigung zu Expansionen erheblich verringern. Im Fokus vieler Untersuchungen steht dabei die Verwendung von Cytosin-Base-Editoren (CBEs), die gezielt Cytosin in Thymin umwandeln können. Im Fall der Huntington-Krankheit ermöglichen sie somit die sichere Mutation von CAG zu CAA innerhalb der Repeats. Dies ist besonders wirkungsvoll, weil CAA als Unterbrechung die Bildung schädlicher DNA-Strukturen verhindert, die für die Expansion der Wiederholungen verantwortlich sind. Studien an Patientenzellen und in entsprechenden Tiermodellen zeigten, dass diese Veränderungen zu einer deutlich reduzierten somatischen Expansion führen können.
So konnten in mit der Pathologie übereinstimmenden Mausmodellen, die mit Adeno-assoziierten Viren (AAV) behandelt wurden, über Wochen nachweislich weniger Expansionen im Gehirn beobachtet werden. Dies deutet darauf hin, dass das Fortschreiten der Erkrankung verlangsamt oder sogar zum Stillstand gebracht werden könnte. Ein weiterer bedeutender Aspekt dieser Therapie ist, dass die vorgenommenen Veränderungen im genetischen Code oft nicht zu einer Änderung der Proteinsequenz führen. Dies minimiert potenzielle Nebenwirkungen durch ungewollte Funktionsverlustmutationen des Huntingtin-Proteins. Zugleich bleibt die Expression des Proteins erhalten, was wichtig ist, da Huntingtin für normale zelluläre Vorgänge essenziell ist.
Durch die Einführung von gleichbedeutenden, synonymen Codonänderungen kann also auf elegante Weise eine Stabilisierung der DNA erzielt werden, ohne die Proteinfunktion zu kompromittieren. Neben den Vorteilen bei der Stabilisierung wurde auch das Risiko unerwünschter Nebenwirkungen eingehend untersucht. Off-Target-Analysen zeigen, dass Base-Editing-Ansätze zwar gewisse, jedoch meist tolerable Nebeneffekte auf nicht-Zielregionen hervorrufen können. Kritisch dabei ist, dass diese unerwünschten Modifikationen hauptsächlich in nichtkodierenden Bereichen der DNA vorkommen und kaum bis selten zu schädlichen Aminosäureänderungen in Proteinen führen. Diese Erkenntnisse sind Grundvoraussetzung für die spätere Entwicklung klinischer Anwendungen, in denen die Sicherheit der Genom-Editierung höchste Priorität hat.
Parallel zu den Untersuchungen an Huntington befassen sich Forscher auch mit ähnlichen Ansätzen für andere trinukleotidische Wiederholungserkrankungen, etwa Friedreich-Ataxie, die durch GAA-Repeats im FXN-Gen verursacht wird. Dabei konnten Adenin-Base-Editoren (ABEs) eingesetzt werden, um GAA-Tripletts zu unterbrechen und so die Wiederholungsstabilität zu erhöhen, was wiederum die Krankheitsprogression verlangsamen kann. Diese konzeptionelle Parallele unterstreicht die allgemeine Anwendbarkeit von Base-Editing-Technologien bei der Behandlung von TNR-Erkrankungen. Ein weiterer interessanter Ansatzpunkt ist die Anwendung speziell entwickelter AAV-Serotypen wie AAV9 für die gezielte Abgabe der Base-Editoren in das Nervengewebe. Durch neonatale Injektionen können hohe Transduktionsraten in neuronalen Zellpopulationen erreicht werden, die von den somatischen Expansionsprozessen besonders betroffen sind.
Diese virale Abgabe gewährleistet eine langanhaltende Expression der Editoren, die zur sukzessiven Einführung von Interruptionsmutationen führt und somit eine nachhaltige Stabilisierung liefert. Die Forschung zeigt auch, dass die Menge der eingebrachten Unterbrechungen mit der Länge der Repeat-Tracts korreliert. Längere, pathogene Repeats bieten mehr Bindungsstellen für den Base-Editor, was höhere Editier-Raten erzeugt. Dieses Phänomen verstärkt den therapeutischen Effekt bei besonders schweren Genotypen. Zudem konnten Behandlungen in Patientenzellen belegen, dass die Interruptionsmutationen zur Erhöhung der normalen Huntingtin-mRNA und Proteinspiegel führen können, was einen zusätzlichen positiven Effekt darstellen könnte.
Eine weitere Herausforderung bei der Genom-Editierung ist die potenzielle Entstehung von Insertions- oder Deletionsmutationen (Indels), die bei Base-Editing-Verfahren deutlich seltener auftreten als bei klassischen CRISPR/Cas9-Methoden. In den beobachteten Systemen wurde gezeigt, dass Frameshifts oder schädliche Indels bei Behandlung minimal sind, wodurch die Integrität des genauen Gens überschaubar beeinflusst wird und die Risiko-Nutzen-Bewertung positiv ausfällt. Auch wenn erste Ergebnisse an Zellkulturen und Mausmodellen vielversprechend sind, fehlt noch der direkte Nachweis, dass diese Editierungen zu einer signifikanten Verbesserung klinischer Symptome führen können. Bekannte Mausmodelle wie Htt.Q111 zeigen nicht alle Eigenschaften der menschlichen Huntington-Erkrankung, insbesondere fehlen oft umfassende motorische oder neurodegenerative Defizite, die bei Patienten deutlich vorhanden sind.
Zukünftige Studien mit fortgeschritteneren Tiermodellen oder sogar klinischen Studien werden zeigen müssen, ob die Vermeidung somatischer Expansionen mittel- bis langfristig einen relevanten Beitrag zur Stabilisierung oder Verbesserung der Erkrankungssituation leistet. Die Entwicklung der Base-Editing-Technologien, insbesondere die Optimierung der Deaminase-Enzyme und der Cas-Oberflächenmoleküle, erlaubt zunehmend gezieltere und wirkungsvollere Eingriffe. Die Kombination aus hoher Zielgenauigkeit, reduziertem Off-Target-Potenzial und schonender DNA-Bearbeitung ist ein entscheidender Fortschritt, der diesen Ansatz auch für die Huntington-Therapie attraktiv macht. Für Patienten, Familien und Mediziner bieten diese Erkenntnisse Hoffnung auf Therapien, die nicht nur Symptome lindern, sondern die erbliche Ursache der Krankheit direkt beeinflussen. Die Grundlage für eine solche Möglichkeiten geben die neuen Belege, dass gezielte Interruptionsmutationen in HTT-Genen die somatische Erweiterung effektiv hemmen und bereits bestehende Expansionen teilweise gar zurückführen können.
Dies kann die bisher als unumstößlich geltende Pathogenese von Huntington potenziell modifizieren. Neben der klinischen Entwicklung müssen ethische und regulatorische Fragen sorgfältig diskutiert werden. Die dauerhafte genetische Veränderung vor allem in Nervenzellen verlangt höchste Sicherheitsstandards und umfassende Aufklärung der Patienten. Die Weiterentwicklung moderner Liefermethoden, etwa auf viraler Basis oder durch virale Vektoren, stellt dabei eine wichtige Grundlage dar. Darüber hinaus steht die Forschung vor der weiteren Aufgabe, die optimale zeitliche Behandlung festzulegen.
Frühzeitige Intervention, idealerweise vor Ausbruch der ersten neurologischen Symptome, könnte den größten Nutzen erzielen, während spätere Behandlungen möglicherweise das Fortschreiten nur noch begrenzt aufhalten. Die genaue Bestimmung der therapeutischen Fenster sowie die individualisierte Anpassung in Abhängigkeit von Gentyp und Krankheitsverlauf sind daher wichtige Forschungsaspekte. Insgesamt stellen die Fortschritte bei der Beeinflussung somatischer Expansionen durch präzise Base Editing einen bedeutenden Meilenstein in der Huntington-Forschung dar. Sie eröffnen neue Wege, die bislang unaufhaltsame genetische Erkrankung zu modifizieren. Die Kombination aus Genetik, molekularer Biologie und fortschrittlichen Biotechnologien steht jetzt vor der Herausforderung, diese Erkenntnisse in wirkungsvolle und sichere Therapien für Betroffene umzusetzen.
Die Zukunft der Behandlung von Huntington könnte somit endgültig einen Schritt näher an eine genetische Korrektur rücken.