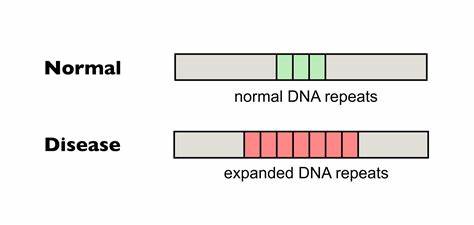
Die Huntington-Krankheit (HD) ist eine erbliche neurodegenerative Erkrankung, die durch erweitere Trinukleotidwiederholungen des CAG-Motivs im Huntingtin-Gen (HTT) ausgelöst wird. Diese sich wiederholenden DNA-Sequenzen können somatisch instabil sein, das heißt sie neigen besonders im Gehirn dazu, mit der Zeit weiter auszudehnen. Diese somatische Expansion ist eng mit der Verschlechterung der klinischen Symptome verbunden und wirkt als kinetisches Damoklesschwert, das den Krankheitsverlauf erschwert. Forschungen zeigen, dass die Länge der reinen, ununterbrochenen CAG-Repeats eine Schlüsselrolle für das Alter beim Krankheitsbeginn und den Schweregrad der Symptome spielt. Je länger die ununterbrochene Repeatlänge, desto schwerwiegender und früher tritt die Krankheit auf.
Die Designierungsmöglichkeiten für die Behandlung haben sich durch innovative genome-editierende Methoden erweitert, insbesondere durch sogenannte Basen-Editoren, die gezielt einzelne Nukleotide verändern können, ohne den DNA-Strang zu brechen. Einer der neuesten Fortschritte in diesem Bereich ist die Verwendung von cytosin- und adenin-Basenediting, um in patienteneigenen Zellen und in Mausmodellen gezielt Unterbrechungen in den Repeat-Trakten einzuführen und somit die Instabilität der sich wiederholenden Sequenzen maßgeblich zu reduzieren. Traditionelle Gen-Editing-Verfahren wie CRISPR/Cas9 generieren Doppelstrangbrüche, deren Reparatur potenziell unerwünschte Einschnitte oder größere Mutationen begünstigen kann. Basen-Editing hingegen wirkt präzise und schonend, indem es einzelne Basen paarspezifisch ändert und so natürliche, stabile Varianten der Wiederholungen nachahmt. Diese sogenannten „Interruptions“ – beispielsweise der Austausch eines CAG-Codons gegen CAA, das ebenfalls Glutamin kodiert – erhöhen die Stabilität der DNA-Sequenz und hemmen die Expansion in somatischen Geweben.
Neuere Studien konnten in patientenabgeleiteten Fibroblasten sowie in Htt.Q111-Mausmodellen zeigen, dass durch eine gezielte cytosinbasierte Umwandlung der CAG-Wiederholungen eine deutliche Verringerung der somatischen Expansion erreicht werden kann. In präklinischen Experimenten konnten mehr als 60 Prozent der HTT-Allele mit synthetisch eingeführten Interruptions modifiziert und die somatische Repeat-Expansion effektiv gebremst werden. Im Verlauf von mindestens 30 Tagen in Kultur zogen sich die Repeatlängen sogar zurück, was auf eine Stabilisierung und potenzielle Reparatur der pathogenen Expansion hinweist. Diese kühnen Schritte eröffnen ein ganz neues Fenster für therapeutische Interventionsstrategien in Huntington und anderen polyglutaminopathischen Erkrankungen, die alle durch ähnliche sich erweiternde CAG-Repeatabschnitte gekennzeichnet sind.
Das Einbringen dieser Interruptions steht im Zusammenhang mit einer Änderung der DNA-Struktur. Ununterbrochene CAG-Stretches neigen dazu, bestimmte höhergeordnete Strukturen wie Haarnadelschleifen, R-Loops und andere nicht-kanonische DNA-Konformationen auszubilden, die eine schlechte Vorlage für die DNA-Reparatursysteme darstellen. Diese Fehlreparaturen begünstigen die Erweiterung der Repeatlänge. Interruptions wiederum schwächen diese Strukturen und erhöhen die DNA-Stabilität, was den Fehlern der Reparaturmechanismen entgegenwirkt. Neben der direkten Manipulation des Huntington-Repeat-Locus konnten Forscher auch die Auswirkungen des somatischen Instabilitätsmanagements auf die Symptomatik und das Fortschreiten der Erkrankung abschätzen.
Das Verhindern oder Reduzieren der somatischen Expansion im Gehirn könnte den Zeitpunkt des Symptombeginns verzögern oder das Fortschreiten verlangsamen. Aktuelle tierexperimentelle Daten zeigen, dass nach der Behandlung mit Adeno-assoziierten Viren (AAVs), die Basen-Editoren trugen, eine signifikante Reduktion der Somanik im Striatum und Kortex möglich ist. Hierbei konnten NIH-3T3 Mäuse effiziente und dauerhafte Editierungen im HTT-Gen mit Bis-Adeninnukleotiden einführen, die für den menschlichen Allelcharakter typisch sind. Die AAV9-abhängige Verabreichung in der frühen Neonatalphase hat sich als besonders wirkungsvoll erwiesen. Der epigenetische und molekulare Reifezustand der Zielzellen spielt hierbei ebenfalls eine Rolle, wobei sich bereits frühe Interventionen als effektiver darstellen.
Auf der Ebene der Sicherheit hat die Untersuchung von Off-Target-Effekten hohe Priorität. Basen-Editoren können theoretisch an ähnlichen Sequenzen ungewollte Veränderungen auslösen, besonders in genomweiten Repeats. Umfangreiche Sequenzanalysen bestätigten allerdings, dass der Großteil der Editierungen an nicht-kodierenden oder synonym bleibenden Regionen auftritt, die keine wesentlichen negativen Effekte auf Proteine haben. Zudem wurde ein Zusammenhang zwischen der Anzahl der Abweichungen der Zielsequenz und der reduzierten Off-Target-Aktivität nachgewiesen. Die Forschung nutzt zudem optimierte Cas9-Varianten mit erweiterten oder alternativen PAM-Spezifitäten, um die Präzision zu steigern und ungewollte Bindungen an falschen Genomstellen zu minimieren.
Im Bereich der Anwendungsmöglichkeiten zeigt sich auch die Strategie des Adenin-Basen-Editings in gleicher Weise vielversprechend, etwa zur Behandlung von Friedreich-Ataxie, einer weiteren TNR-Erkrankung, die durch Expansion von GAA-Trinukleotiden im FXN-Gen verursacht wird. Die Prinzipien der Reduktion somatischer Repeat-Erweiterungen durch gezielte Unterbrechungen lassen sich dabei übertragen. Klinische Übersetzung und Entwicklung Wir befinden uns aktuell in der präklinischen Phase, in der Wirksamkeit und Sicherheit in Zellkulturen und Tiermodellen evaluiert werden. Die Entwicklung geeigneter AAV-Vektoren, die breite und gezielte Transduktion von relevanten Hirnregionen ermöglichen, etwa dem Striatum, dem Kortex und weiteren Kernbereichen, steht im Fokus. Die langfristige Expression von Basen-Editoren in vivo stellt sowohl eine Chance als auch ein Risiko dar.
So ermöglicht die konstante Anwesenheit des Editors eine schrittweise Anreicherung der Interruptions in den Repeat-Trakten, andererseits könnte es zu kumulativen Off-Target-Effekten kommen. Forschungseinrichtungen und Unternehmen erforschen daher alternative Liefermethoden, etwa transientes mRNA-Delivery oder Virus-ähnliche Partikel, um das Risiko zu minimieren und die Behandlung sicherer zu gestalten. Vital ist ebenso die zeitliche Feinabstimmung der Intervention, da die somatische Expansion oft über Jahrzehnte stillschweigend stattfindet, bevor neurologische Symptome auftreten. Frühzeitige Eingriffe, möglicherweise vor dem klinischen Beginn der Erkrankung, könnten daher den größten therapeutischen Nutzen erzielen. Zusätzlich wird die Rolle der Interruptions im familiären Kontext diskutiert: Studien zeigen, dass betroffene Familien mit Interruptions in ihren Repeat-Trakten eine spürbar spätere Erkrankung manifestieren oder mildere Verläufe haben.
Daraus lässt sich ableiten, dass die künstliche Einbringung solcher Unterbrechungen nicht nur die somatische Instabilität verringern, sondern auch den generationsübergreifenden Repeat-Transfer beeinflussen und verlangsamen könnte. Ausblick Aus heutiger Sicht eröffnen präzise Gen-Editierungstechnologien eine neue Ära in der Behandlung von Huntington und verwandten TNR-Erkrankungen. Die Möglichkeit, krankheitsverursachende Repeat-Expansions zu stabilisieren oder gar zurückzudrängen, stellt einen Paradigmenwechsel dar. In Zukunft könnten diese Therapien als monotherapeutische Ansätze oder in Kombination mit symptomatisch wirksamen Medikamenten eingesetzt werden. Entscheidend wird die weitere Optimierung von Editoren, die Minimierung von Nebenwirkungen sowie die Etablierung zuverlässiger, sicherer und effizienzbasierter Lieferwege sein.
Ebenso wichtig sind klinische Studien, die zeigen, wie gut sich das molekulare Korrelat der somatischen Repeat-Expansion mit klinischen Ergebnissen verbinden lässt. Die wissenschaftlichen Fortschritte im Bereich der Basen-Editierung liefern einen wegweisenden Ansatz, der letztlich das Leben von Patienten mit Huntington und anderen TNR-Erkrankungen erheblich verbessern könnte.